欢迎访问《中国农学通报》,
中国农学通报 ›› 2026, Vol. 42 ›› Issue (6): 54-63.doi: 10.11924/j.issn.1000-6850.casb2025-0463
罗燕羽( ), 魏利国, 刘伟光, 杨志建, 张进忠, 陈继玮, 黄绍力()
), 魏利国, 刘伟光, 杨志建, 张进忠, 陈继玮, 黄绍力()
收稿日期:2025-06-21
修回日期:2025-10-11
出版日期:2026-03-25
发布日期:2026-03-30
通讯作者:
作者简介:罗燕羽,女,1993年出生,福建漳州人,农艺师,硕士,研究方向:植物新品种选育、组培快繁及栽培技术研究。通信地址:510335 广州市海珠区琶洲大道17-19号广州市农业农村科学院,Tel:020-84215921,E-mail:874940453@qq.com。
基金资助:
LUO Yanyu(), WEI Liguo, LIU Weiguang, YANG Zhijian, ZHANG Jinzhong, CHEN Jiwei, HUANG Shaoli()
Received:2025-06-21
Revised:2025-10-11
Published:2026-03-25
Online:2026-03-30
摘要:
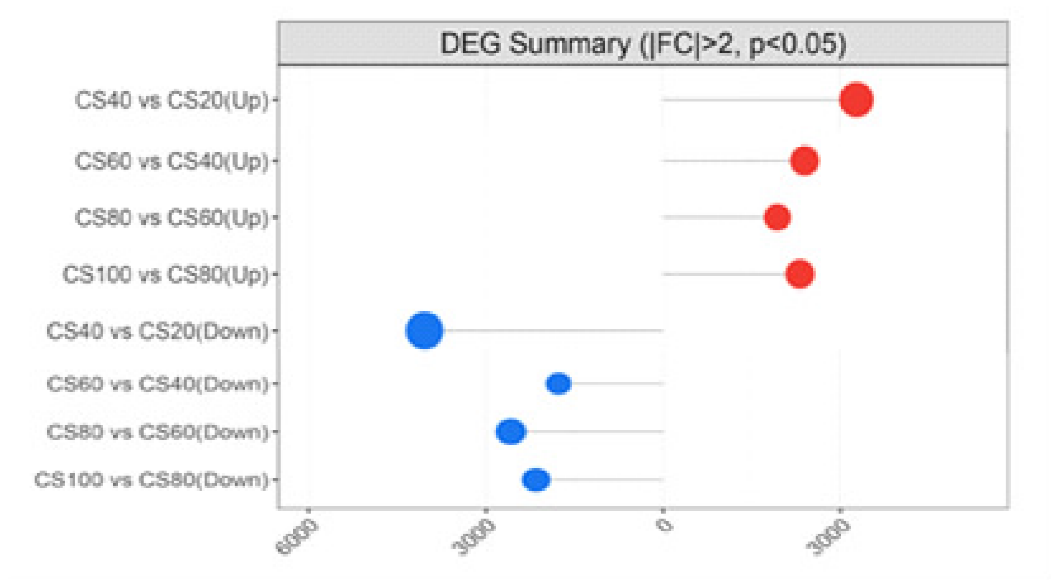
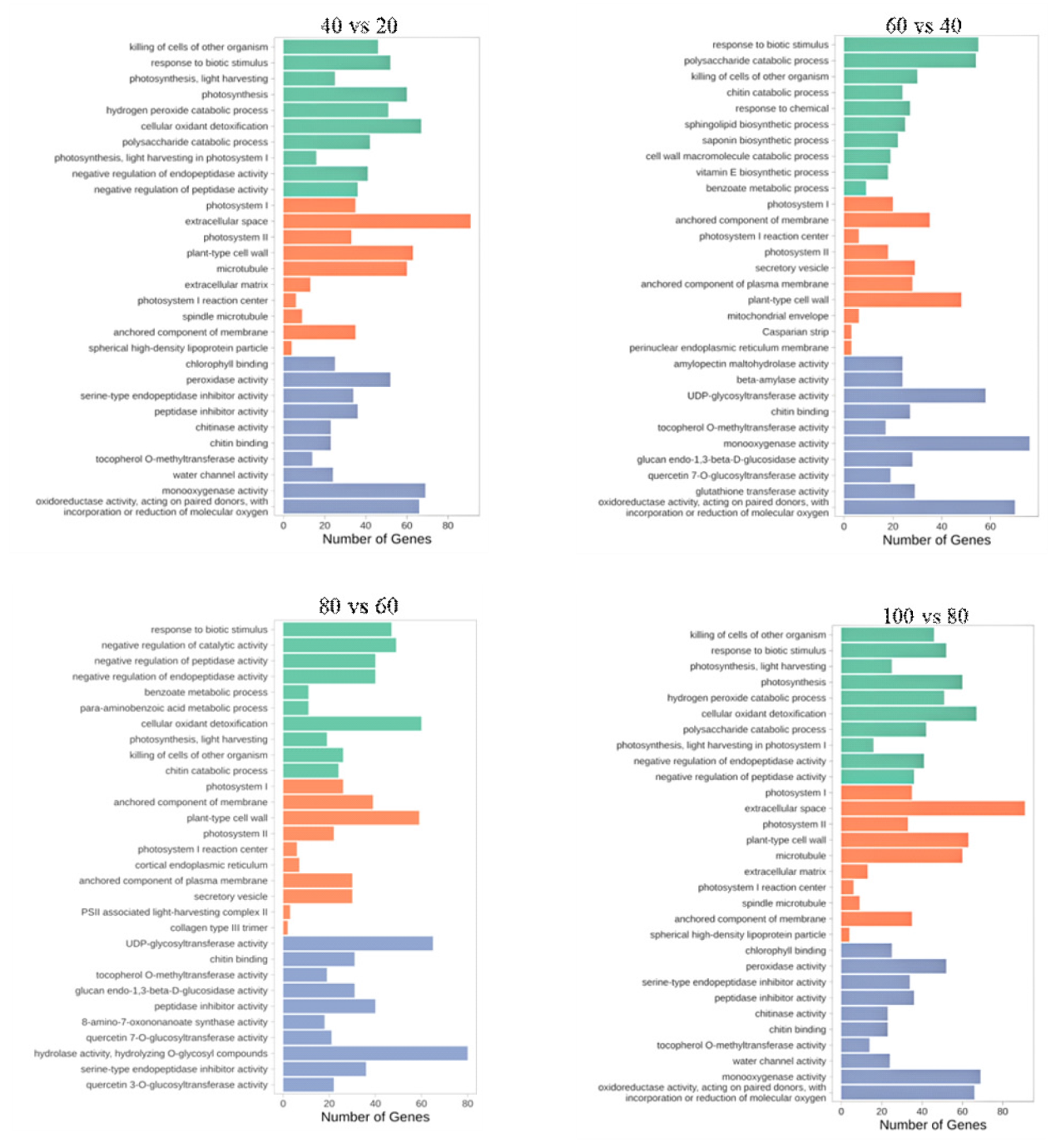
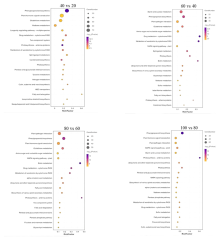
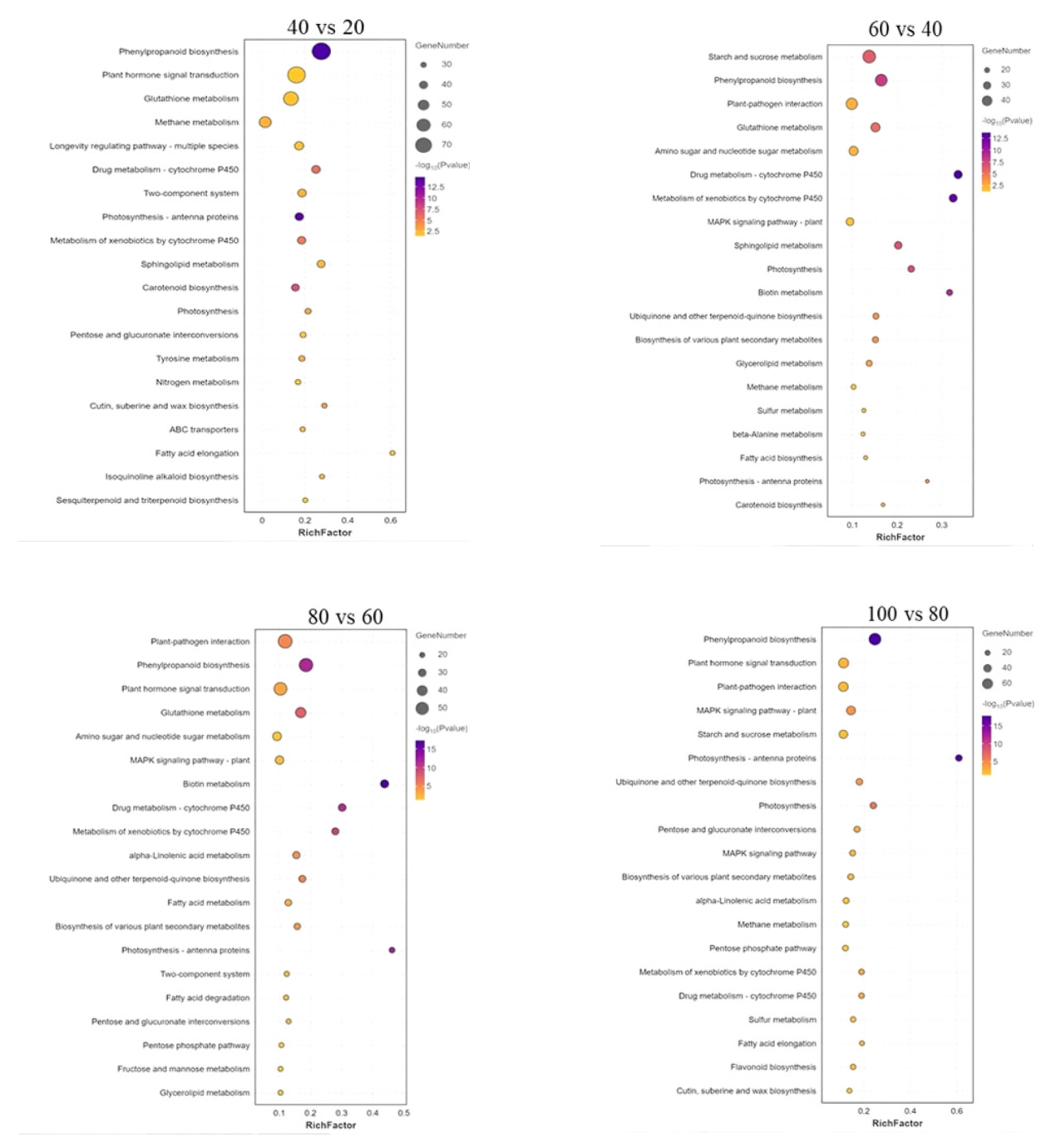
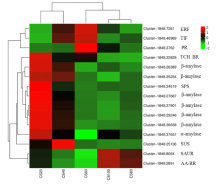
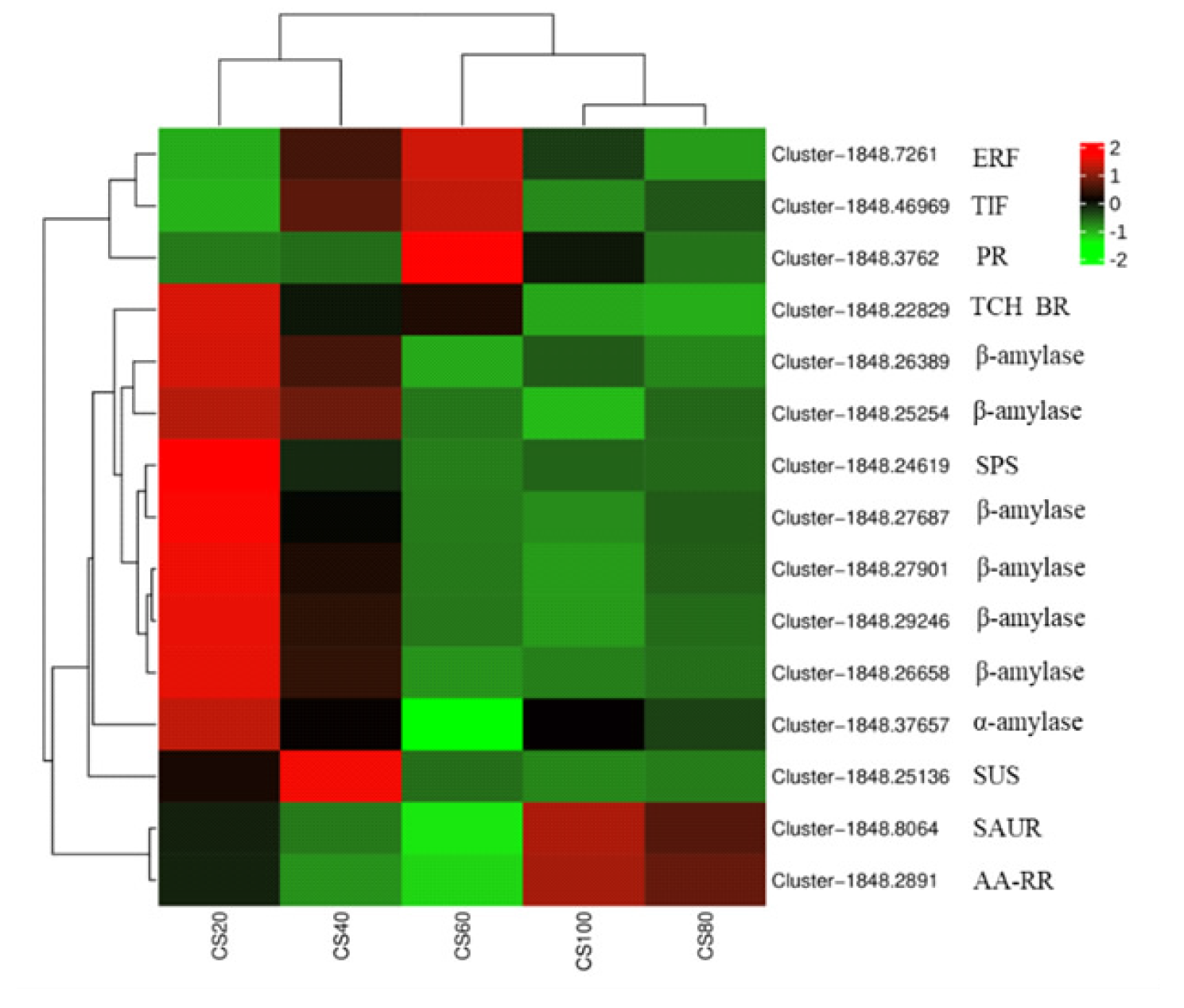
为探究槟榔香芋块茎发育的相关机理,挖掘与块茎发育相关的关键基因,本研究以广东炭步文冈槟榔香芋单株试管苗为试验材料,对不同蔗糖浓度处理的试管芋块茎进行转录组测序分析,筛选块茎发育相关差异表达基因,并采用实时荧光定量PCR (qRT-PCR)技术对候选基因进行表达验证。试验结果表明,4个比较组共有89320个unigene,差异表达基因共20458个,其中上调基因9925个,下调基因10533个。通过GO富集分析发现,这些差异基因主要富集在生物过程,特别是对生物刺激的响应以及细胞氧化解毒等方面;KEGG富集分析结果表明,这些差异基因富集的通路主要在苯丙素生物合成、植物激素信号转导、淀粉和蔗糖代谢以及植物病原互作等通路。此外,本研究从淀粉和蔗糖代谢、植物激素信号转导通路中筛选出15个核心候选基因,经qRT-PCR验证的6个基因,其表达模式与转录组测序结果基本一致。本研究结果为芋块茎膨大的分子机制研究提供了重要数据支撑,也为进一步解析槟榔香芋块茎发育的调控网络奠定了理论基础。
罗燕羽, 魏利国, 刘伟光, 杨志建, 张进忠, 陈继玮, 黄绍力. 蔗糖诱导槟榔香芋试管芋形成的转录组分析及关键基因的筛选[J]. 中国农学通报, 2026, 42(6): 54-63.
LUO Yanyu, WEI Liguo, LIU Weiguang, YANG Zhijian, ZHANG Jinzhong, CHEN Jiwei, HUANG Shaoli. Transcriptome Analysis of Sucrose Induced Areca Taro in Vitro and Key Genes Screening[J]. Chinese Agricultural Science Bulletin, 2026, 42(6): 54-63.
| 基因 | 序列 | |
|---|---|---|
| Actin | F: | CTAGTGGTCGCACAACAGGT |
| R: | TTCACGCTCAGCAGTGGTAG | |
| Cluster-1848.46969 | F: | CCTCACCATCTTCTACAATGGCAC |
| R: | GTGATCCCTGTCTCCACCATCTTA | |
| Cluster-1848.25136 | F: | AGCTGGATTTCGAGCCCTTC |
| R: | GCCTTTGCCAAACTGGACTG | |
| Cluster-1848.25254 | F: | GCGAGGGCCCCAAGATTTAT |
| R: | ATCATCCCCAACGTTCCCAC | |
| Cluster-1848.24619 | F: | CTCGCAAGACGAAGCAGGTC |
| R: | GCATTCAAATAAAGCAGGAACCCC | |
| Cluster-1848.7261 | F: | GAACGACTCGGAGGAGATGCT |
| R: | ATCTTCCCTGCTCTTCGACTCC | |
| Cluster-1848.26389 | F: | CTGGGATGGTGACAGACATAGAGG |
| R: | ATTCCCCTGAAATTCTCCAATGCC | |
| 基因 | 序列 | |
|---|---|---|
| Actin | F: | CTAGTGGTCGCACAACAGGT |
| R: | TTCACGCTCAGCAGTGGTAG | |
| Cluster-1848.46969 | F: | CCTCACCATCTTCTACAATGGCAC |
| R: | GTGATCCCTGTCTCCACCATCTTA | |
| Cluster-1848.25136 | F: | AGCTGGATTTCGAGCCCTTC |
| R: | GCCTTTGCCAAACTGGACTG | |
| Cluster-1848.25254 | F: | GCGAGGGCCCCAAGATTTAT |
| R: | ATCATCCCCAACGTTCCCAC | |
| Cluster-1848.24619 | F: | CTCGCAAGACGAAGCAGGTC |
| R: | GCATTCAAATAAAGCAGGAACCCC | |
| Cluster-1848.7261 | F: | GAACGACTCGGAGGAGATGCT |
| R: | ATCTTCCCTGCTCTTCGACTCC | |
| Cluster-1848.26389 | F: | CTGGGATGGTGACAGACATAGAGG |
| R: | ATTCCCCTGAAATTCTCCAATGCC | |
| 样本 | 原始碱基总数/bp | 过滤后碱基总数/bp | Q20/% | Q30/% | GC/% |
|---|---|---|---|---|---|
| CS20-1 | 6,133,285,800 | 6,073,890,927 | 98.39 | 95.05 | 53.08 |
| CS20-2 | 5,670,601,200 | 5,615,668,581 | 98.21 | 94.56 | 52.95 |
| CS20-3 | 6,634,682,700 | 6,565,846,280 | 98.17 | 94.43 | 52.92 |
| CS40-1 | 6,999,130,200 | 6,921,367,632 | 98.16 | 94.38 | 53.94 |
| CS40-2 | 6,999,735,300 | 6,930,637,562 | 98.48 | 95.19 | 54.02 |
| CS40-3 | 6,462,959,700 | 6,390,142,404 | 98.04 | 94.05 | 53.54 |
| CS60-1 | 6,614,923,500 | 6,539,947,082 | 98.34 | 94.80 | 53.04 |
| CS60-2 | 6,896,487,300 | 6,811,281,892 | 98.10 | 94.20 | 53.03 |
| CS60-3 | 7,000,299,300 | 6,926,033,621 | 98.21 | 94.50 | 53.58 |
| CS80-1 | 5,530,323,300 | 5,468,534,702 | 98.09 | 94.17 | 52.68 |
| CS80-2 | 6,117,102,000 | 6,051,141,458 | 98.13 | 94.22 | 52.52 |
| CS80-3 | 6,175,827,300 | 6,110,304,708 | 98.16 | 94.31 | 52.56 |
| CS100-1 | 6,743,600,700 | 6,678,423,830 | 98.38 | 94.96 | 54.25 |
| CS100-2 | 5,594,808,600 | 5,538,444,424 | 98.29 | 94.73 | 53.99 |
| CS100-3 | 5,833,727,700 | 5,767,373,503 | 98.09 | 94.23 | 53.87 |
| 样本 | 原始碱基总数/bp | 过滤后碱基总数/bp | Q20/% | Q30/% | GC/% |
|---|---|---|---|---|---|
| CS20-1 | 6,133,285,800 | 6,073,890,927 | 98.39 | 95.05 | 53.08 |
| CS20-2 | 5,670,601,200 | 5,615,668,581 | 98.21 | 94.56 | 52.95 |
| CS20-3 | 6,634,682,700 | 6,565,846,280 | 98.17 | 94.43 | 52.92 |
| CS40-1 | 6,999,130,200 | 6,921,367,632 | 98.16 | 94.38 | 53.94 |
| CS40-2 | 6,999,735,300 | 6,930,637,562 | 98.48 | 95.19 | 54.02 |
| CS40-3 | 6,462,959,700 | 6,390,142,404 | 98.04 | 94.05 | 53.54 |
| CS60-1 | 6,614,923,500 | 6,539,947,082 | 98.34 | 94.80 | 53.04 |
| CS60-2 | 6,896,487,300 | 6,811,281,892 | 98.10 | 94.20 | 53.03 |
| CS60-3 | 7,000,299,300 | 6,926,033,621 | 98.21 | 94.50 | 53.58 |
| CS80-1 | 5,530,323,300 | 5,468,534,702 | 98.09 | 94.17 | 52.68 |
| CS80-2 | 6,117,102,000 | 6,051,141,458 | 98.13 | 94.22 | 52.52 |
| CS80-3 | 6,175,827,300 | 6,110,304,708 | 98.16 | 94.31 | 52.56 |
| CS100-1 | 6,743,600,700 | 6,678,423,830 | 98.38 | 94.96 | 54.25 |
| CS100-2 | 5,594,808,600 | 5,538,444,424 | 98.29 | 94.73 | 53.99 |
| CS100-3 | 5,833,727,700 | 5,767,373,503 | 98.09 | 94.23 | 53.87 |
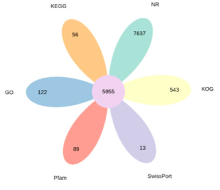
| 数据库 | 基因数目 | 占比/% |
|---|---|---|
| NR | 46769 | 52.36 |
| KOG | 27192 | 30.44 |
| SwissPort | 22292 | 24.96 |
| Pfam | 24459 | 27.38 |
| GO | 34176 | 38.26 |
| KEGG | 12387 | 13.87 |
| 在所有数据库中得到注释 | 5955 | 6.67 |
| 至少在一个数据库中得到注释 | 48058 | 53.80 |
| 总基因 | 89320 | 100.00 |
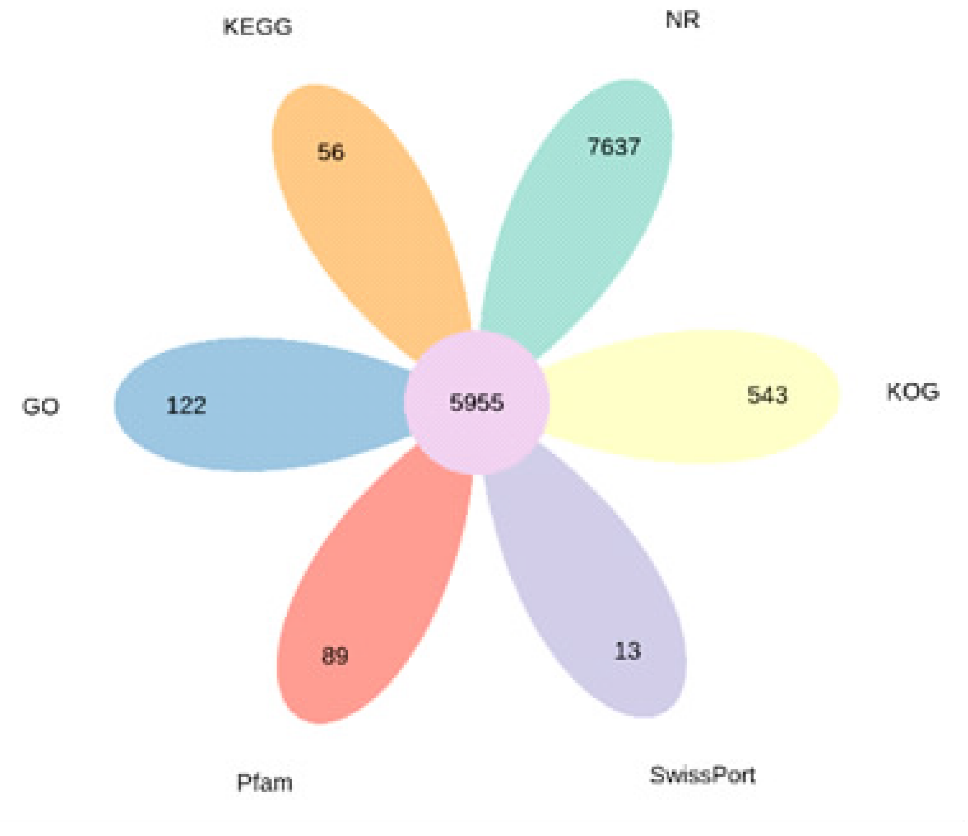
| 数据库 | 基因数目 | 占比/% |
|---|---|---|
| NR | 46769 | 52.36 |
| KOG | 27192 | 30.44 |
| SwissPort | 22292 | 24.96 |
| Pfam | 24459 | 27.38 |
| GO | 34176 | 38.26 |
| KEGG | 12387 | 13.87 |
| 在所有数据库中得到注释 | 5955 | 6.67 |
| 至少在一个数据库中得到注释 | 48058 | 53.80 |
| 总基因 | 89320 | 100.00 |
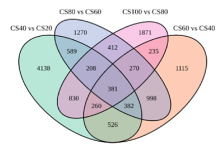
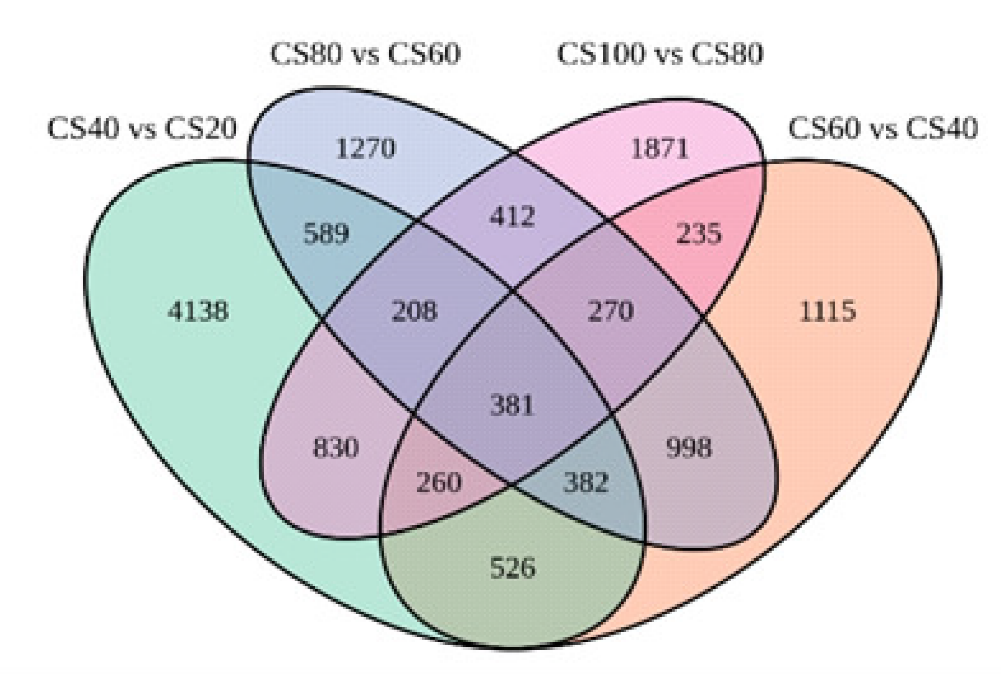
| 比较组 | 通路数量 | 总差异基因 | 上调基因 | 下调基因 |
|---|---|---|---|---|
| 40 VS 20 | 36 | 1329 | 696 | 633 |
| 60 VS 40 | 40 | 806 | 444 | 362 |
| 80 VS 60 | 47 | 780 | 314 | 466 |
| 100 VS 80 | 61 | 958 | 589 | 369 |
| 比较组 | 通路数量 | 总差异基因 | 上调基因 | 下调基因 |
|---|---|---|---|---|
| 40 VS 20 | 36 | 1329 | 696 | 633 |
| 60 VS 40 | 40 | 806 | 444 | 362 |
| 80 VS 60 | 47 | 780 | 314 | 466 |
| 100 VS 80 | 61 | 958 | 589 | 369 |
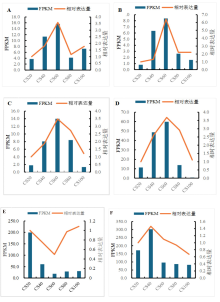
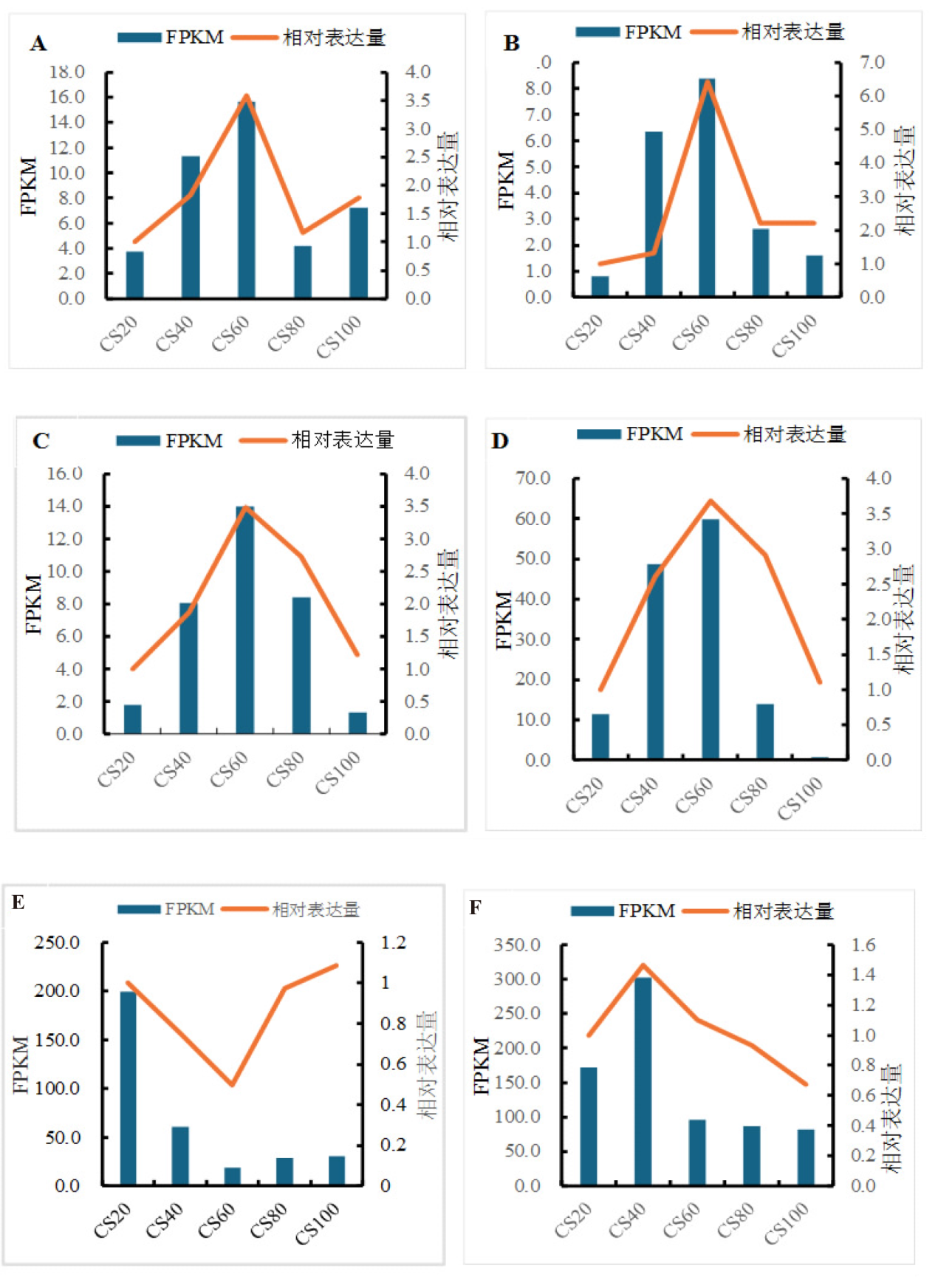
| [1] |
于继英, 田子罡, 徐美娟, 等. 我国芋头资源分布和饲用情况[J]. 当代畜牧, 2018(33):22-26.
|
| [2] |
|
| [3] |
戴修纯, 罗燕羽, 黄绍力, 等. 广东省芋头产业现状与发展对策[J]. 广东农业科学, 2021, 48(6):126-135.
|
| [4] |
张雨濛. 芋球茎淀粉积累与食味品质形成的关系及其对5-ALA响应研究[D]. 扬州: 扬州大学, 2022.
|
| [5] |
谭卫萍, 许敏娜, 肖熙鸥. 广州市文冈香芋发展现状与应对策略[J]. 南方农业, 2011, 5(5):77-79.
|
| [6] |
杨璐, 刘希元, 张广臣. 光周期、蔗糖及6-BA对马铃薯试管薯诱导的影响[J]. 吉林农业大学学报, 2022, https://link.cnki.net/urlid/22.1100.S.20220512.1913.004.
|
| [7] |
严华兵, 周慧文, 曾文丹. 木薯试管块根诱导技术研究[J]. 热带作物学报, 2016, 37(9):1708-1713.
|
| [8] |
韩晓勇, 王立, 殷剑美, 等. 靖江香沙芋试管芋诱导影响因素研究[J]. 浙江农业科学, 2017, 58(10):1766-1769.
|
| [9] |
周逊, 徐晓舒, 易迁, 等. 不同浓度蔗糖诱导对试管姜丙二醛含量的影响[J]. 北方园艺, 2013(17):104-106.
|
| [10] |
doi: 10.1006/anbo.1995.1024 URL |
| [11] |
庄赛伟, 刘铭, 梅朋飞, 等. 蔗糖对参薯块茎形成及DamiR156/DaSPL的调控分析[J]. 分子植物育种, 2024, 22(7):2098-2103.
|
| [12] |
doi: 10.1080/01904167.2021.1881545 URL |
| [13] |
卢晓月, 李大伟, 邬倩, 等. 马铃薯块茎不同发育时期转录组分析[J]. 云南师范大学学报(自然科学版), 2021, 41(4):25-32.
|
| [14] |
高小溪. 蔗糖诱导马铃薯(S. tuberosum L.)试管薯形成的转录组分析与关键基因筛选[D]. 武汉: 华中农业大学, 2006.
|
| [15] |
赵令敏. 淀粉-蔗糖代谢对山药(Diocscorea opposita Thunb.)块茎膨大调控机制的研究[D]. 呼和浩特: 内蒙古农业大学, 2023.
|
| [16] |
赵娜, 刘宇曦, 张朝澍, 等. 不同马铃薯淀粉含量差异的转录组学解析[J]. 作物学报, 2024, 50(6):1503-1513.
|
| [17] |
刘星月. 红芽芋脱毒种茎诱导及形成机制初步研究[D]. 南昌: 江西农业大学, 2020.
|
| [18] |
尹明华, 白丽, 陈舒敏, 等. 广丰千金薯和铁棍山药脱毒微型块茎的转录组分析[J]. 浙江农业学报, 2022, 34(10):2209-2219.
doi: 10.3969/j.issn.1004-1524.2022.10.15 |
| [19] |
黄绍力, 罗燕羽, 魏利国, 等. 槟榔芋脱毒试管芋的诱导及种苗繁育研究[J]. 中国瓜菜, 2024, 37(9):109-115.
|
| [20] |
doi: 10.1038/nbt.1883 |
| [21] |
doi: 10.1093/nar/gkn176 pmid: 18445632 |
| [22] |
|
| [23] |
doi: 10.1006/meth.2001.1262 URL |
| [24] |
单建伟, 柳俊, 索海翠, 等. 糖信号调控马铃薯块茎发育的研究进展[J]. 华中农业大学学报, 2021, 40(4):27-35.
|
| [25] |
doi: 10.1093/jxb/ert474 pmid: 24453229 |
| [26] |
|
| [27] |
李慧敏, 梁永书, 南文斌, 等. 糖调控植物根系生长发育的研究进展[J]. 中国农学通报, 2015, 31(14):108-113.
doi: 10.11924/j.issn.1000-6850.casb15010181 |
| [28] |
胡云海, 蒋先明. 不同糖类和BA对马铃薯(S. Tuberosum)试管薯的影响[J]. 马铃薯杂志, 1989, 3(4):203-206.
|
| [29] |
doi: 10.1038/s41598-020-58167-4 pmid: 31980689 |
| [30] |
doi: 10.1016/j.pbi.2004.03.014 URL |
| [31] |
胡云海, 蒋先明. 植物激素对微型薯形成的影响[J]. 马铃薯杂志, 1992, 6(1):14-22.
|
| [32] |
冯璐. 植物生长物质对新品种马铃薯试管苗生长及微型薯诱导的影响[D]. 太原: 山西农业大学, 2017.
|
| [33] |
张飞, 王艳秋, 朱凯, 等. 不同耐盐性高粱在盐逆境下的比较转录组分析[J]. 中国农业科学, 2019, 52(22):4002-4015.
doi: 10.3864/j.issn.0578-1752.2019.22.006 |
| [34] |
杜文丽, 陈中钐, 许端祥, 等. 低温胁迫下苦瓜叶片转录组差异基因分析及生理响应特征[J]. 核农学报, 2021, 35(2):338-348.
doi: 10.11869/j.issn.100-8551.2021.02.0338 |
| [35] |
徐红霞, 周慧芬, 李晓颖, 等. 低温胁迫下枇杷不同发育阶段的花果转录组比较分析[J]. 园艺学报, 2021, 48(9):1680-1694.
doi: 10.16420/j.issn.0513-353x.2021- 0219 |
| [1] | 张鑫, 郭志鸿, 柴娟, 马荣, 周锋, 王琼, 姚卓男. 兰州百合组培增殖系数与鳞茎膨大的关键影响因子研究[J]. 中国农学通报, 2025, 41(31): 41-46. |
| [2] | 何柳, 原小燕, 张玉松, 刘丽晶, 钟丽琼, 赵凯琴, 张立帆, 何晓莹, 符明联. 花生蔗糖含量与气象因素和品质性状的相关性分析[J]. 中国农学通报, 2025, 41(3): 18-24. |
| [3] | 郭春梅, 肖皓钧, 秦兴利, 丁映风, 沈辉, 杜艳萍, 张宏, 杨正安. 配方施肥对设施基质栽培番茄品质及产量的影响[J]. 中国农学通报, 2025, 41(1): 42-47. |
| [4] | 蔡华扬, 于连升, 李腾鑫, 林宜萌, 杜仁鹏. 生物信息学分析葡聚糖蔗糖酶的结构和功能特性[J]. 中国农学通报, 2024, 40(18): 105-114. |
| [5] | 李娜, 张文玉, 孙纲, 宋晗, 胡秀芹, 辛杰, 王振. 植物SnRK2基因家族的研究进展[J]. 中国农学通报, 2023, 39(30): 108-113. |
| [6] | 洪森荣, 朱盈盈, 李紫莹, 胡明艳, 欧阳克蕙. 盐胁迫下金花菜和紫花苜蓿试管苗的转录组分析及其耐盐基因筛选[J]. 中国农学通报, 2023, 39(3): 111-118. |
| [7] | 高文瑞, 孙艳军, 韩冰, 费聪, 王显生, 徐刚. 弱光对西瓜果实品质及蔗糖代谢的影响[J]. 中国农学通报, 2023, 39(1): 56-61. |
| [8] | 刘永惠, 沈一, 沈悦, 梁满, 陈志德. 花生籽仁发育过程中糖分积累特征及蔗糖代谢酶活性分析[J]. 中国农学通报, 2022, 38(30): 29-34. |
| [9] | 郭书亚, 尚赏, 王坤, 汤其宁, 张艳, 付国占, 卢广远. 秸秆覆盖深松对夏玉米田土壤酶活性的影响[J]. 中国农学通报, 2022, 38(25): 96-101. |
| [10] | 赵晶晶, 周浓, 郑殿峰. 低温胁迫对大豆花期叶片蔗糖代谢及产量的影响[J]. 中国农学通报, 2021, 37(9): 1-8. |
| [11] | 王书玉, 黎蓝, 赖凌峰, 李红梅, 刘季平, 何生根. 纳米铜加蔗糖处理对非洲菊切花的保鲜作用[J]. 中国农学通报, 2021, 37(4): 62-67. |
| [12] | 谢洪宝, 于贺, 陈一民, 隋跃宇, 焦晓光. 秸秆深埋对不同氮肥水平土壤蔗糖酶活性的影响[J]. 中国农学通报, 2021, 37(24): 79-83. |
| [13] | 徐若, 张秀芬, 李艳冰, 字淑慧, 杨生超, 刘涛. 干旱胁迫对三七生理指标的影响及转录组分析[J]. 中国农学通报, 2021, 37(16): 51-58. |
| [14] | 朱琳, 白朕卿, 王延峰, 吴佳文. 非生物胁迫对能源作物糖分产量影响的研究进展[J]. 中国农学通报, 2021, 37(10): 6-11. |
| [15] | 宋少帅, 吴凡, 刘会文, 贺小彦, 穆平. 小麦TaPLD-2基因的克隆及功能分析[J]. 中国农学通报, 2020, 36(21): 98-103. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||