欢迎访问《中国农学通报》,
中国农学通报 ›› 2025, Vol. 41 ›› Issue (25): 153-164.doi: 10.11924/j.issn.1000-6850.casb2025-0057
• 水产·渔业 • 上一篇
郑昀凯( ), 刘鹏辉, 牛世洋, 杨天燕()
), 刘鹏辉, 牛世洋, 杨天燕()
收稿日期:2025-01-16
修回日期:2025-06-03
出版日期:2025-09-05
发布日期:2025-09-16
通讯作者:
作者简介:郑昀凯,男,2003年出生,浙江衢州人,研究方向:海洋资源与环境。通信地址:316022 浙江省舟山市定海区临城街道海大南路1号,E-mail:1257263225@qq.com。
基金资助:
ZHENG Yunkai(), LIU Penghui, NIU Shiyang, YANG Tianyan()
Received:2025-01-16
Revised:2025-06-03
Published:2025-09-05
Online:2025-09-16
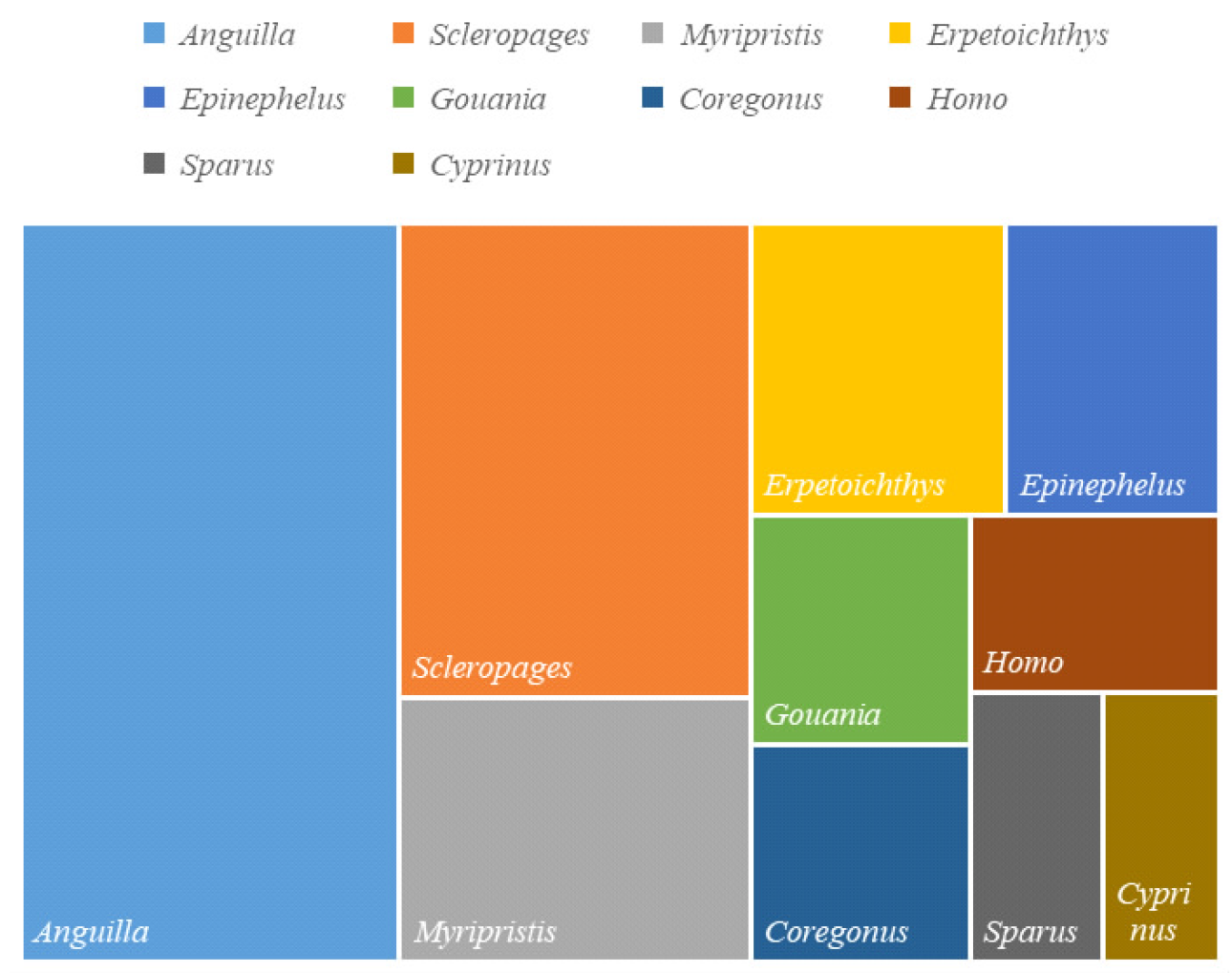
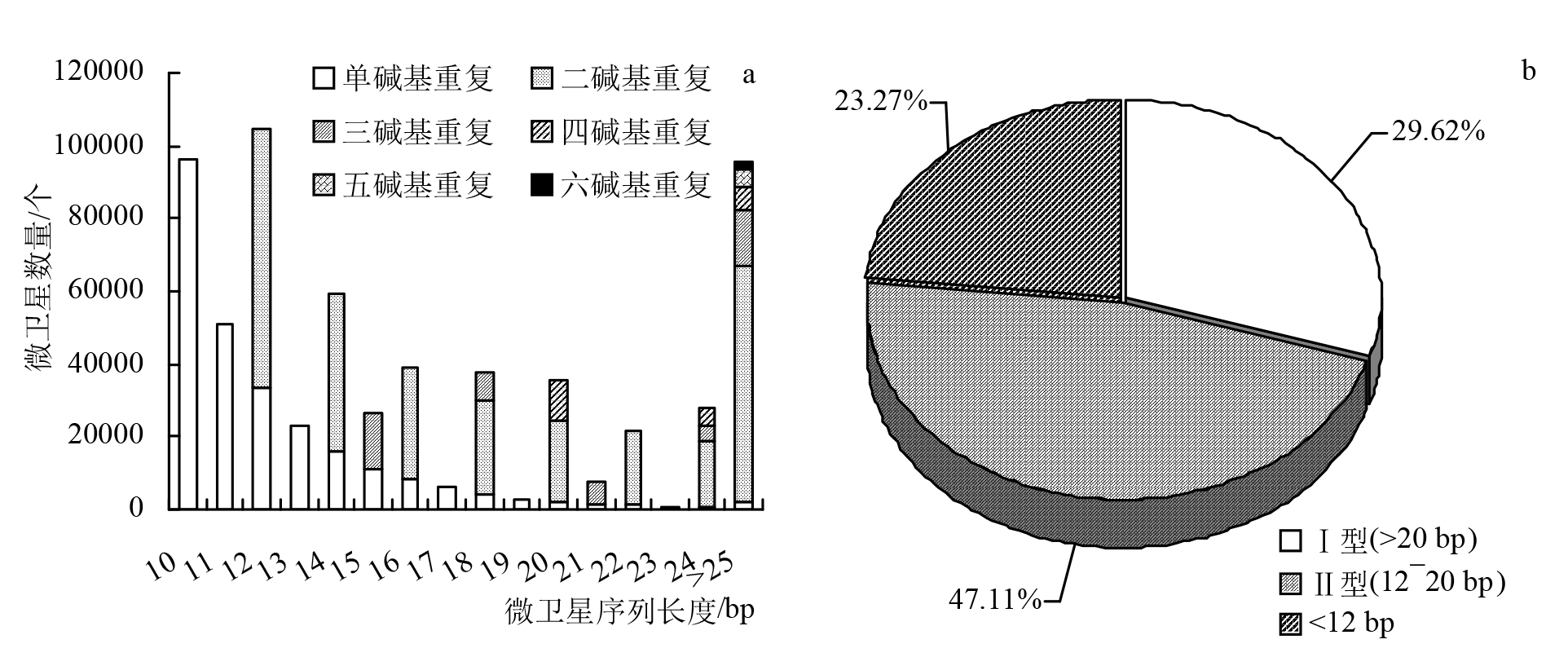
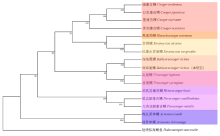
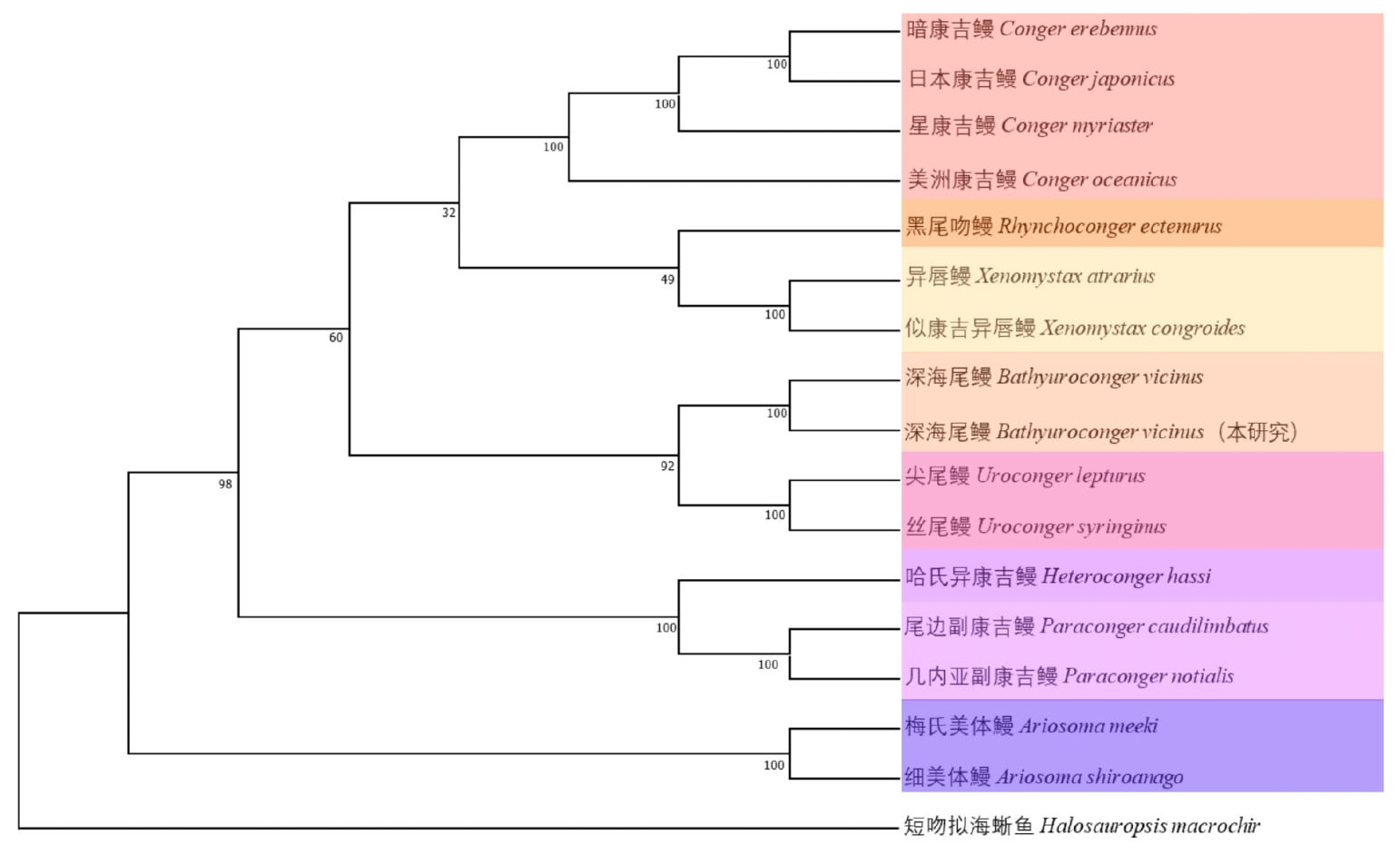
摘要: 为探究深海尾鳗对极端环境的基因组适应机制及系统演化地位,本研究首次对其开展全基因组Survey测序与系统分析。采用Illumina NovaSeq 6000平台进行基因组测序,结合SOAPdenovo2组装基因组草图,利用MISA筛选微卫星位点,基于线粒体蛋白质编码基因串联序列构建系统发育树。结果显示,深海尾鳗基因组大小约为1414 Mb,杂合率、重复序列比例分别为0.66%、54.93%。筛选得到总长度为10936848 bp的微卫星位点共计1221434个,分布在698950条序列上,发生频率、出现频率和相对丰度分别为19.18%、33.53%和857.57个/Mb。在6种完整型微卫星中,二碱基重复类型占比最大(804493个,65.86%)、出现频率最高(22.08%)且相对丰度最大(564.84个/Mb)。在1484种微卫星重复基元中,种类最多的为六碱基重复基元(668种),其次为五碱基重复(583种)、四碱基重复(231种)、三碱基重复(60种)、二碱基重复(12种)和单碱基重复(4种)。其优势碱基类别分别为A(128843,43.40%)、CA(239966,29.83%)、AAT(9533,12.30%)、AAAT(2663,8.16%)、CATTA(316,5.40%)和CACACT(268,6.56%)。基于线粒体蛋白质编码基因串联序列构建的最大似然树显示,深海尾鳗与尖尾鳗属鱼类的亲缘关系较近。本研究揭示了深海尾鳗基因组的复杂特征及丰富的多态性微卫星位点,为其深海适应机制研究提供了分子基础;筛选的微卫星位点可用于种群遗传学分析;系统发育结果支持其与尖尾鳗属鱼类的近缘关系,明确了其在康吉鳗亚科中的演化地位,并提示康吉鳗科可能为多系群。
郑昀凯, 刘鹏辉, 牛世洋, 杨天燕. 基于基因组Survey数据的深海尾鳗微卫星特征及系统发育关系分析[J]. 中国农学通报, 2025, 41(25): 153-164.
ZHENG Yunkai, LIU Penghui, NIU Shiyang, YANG Tianyan. Analysis of Microsatellites Characterization and Phylogenetic Relationship for Bathyuroconger vicinus Based on Genomic Survey Data[J]. Chinese Agricultural Science Bulletin, 2025, 41(25): 153-164.
| 亚科 | 属 | 种 | GenBank序列号 |
|---|---|---|---|
| 深海康吉鳗亚科Bathymyrinae | 美体鳗属Ariosoma | 梅氏美体鳗Ariosoma meeki | OK585090 |
| 细美体鳗Ariosoma shiroanago | AP010861 | ||
| 副康吉鳗属Paraconger | 几内亚副康吉鳗Paraconger notialis | AP010860 | |
| 尾边副康吉鳗Paraconger caudilimbatus | OR582666 | ||
| 异康吉鳗亚科Heterocongrinae | 异康吉鳗属Heteroconger | 哈氏异康吉鳗Heteroconger hassi | AP010859 |
| 康吉鳗亚科Congrinae | 康吉鳗属Conger | 暗康吉鳗Conger erebennus | OM691699 |
| 星康吉鳗Conger myriaster | AB038381 | ||
| 美洲康吉鳗Conger oceanicus | OR546244 | ||
| 日本康吉鳗Conger japonicus | KR131863 | ||
| 异唇鳗属Xenomystax | 似康吉异唇鳗Xenomystax congroides | OR482507 | |
| 异唇鳗Xenomystax atrarius | OR482465 | ||
| 吻鳗属Rhynchoconger | 黑尾吻鳗Rhynchoconger ectenurus | MN256490 | |
| 尖尾鳗属Uroconger | 尖尾鳗Uroconger lepturus | OR581023 | |
| 丝尾鳗Uroconger syringinus | OP056976 | ||
| 深海尾鳗属Bathyuroconger | 深海尾鳗Bathyuroconger vicinus | OP035192 | |
| 深海尾鳗Bathyuroconger vicinus | 本研究 |
| 亚科 | 属 | 种 | GenBank序列号 |
|---|---|---|---|
| 深海康吉鳗亚科Bathymyrinae | 美体鳗属Ariosoma | 梅氏美体鳗Ariosoma meeki | OK585090 |
| 细美体鳗Ariosoma shiroanago | AP010861 | ||
| 副康吉鳗属Paraconger | 几内亚副康吉鳗Paraconger notialis | AP010860 | |
| 尾边副康吉鳗Paraconger caudilimbatus | OR582666 | ||
| 异康吉鳗亚科Heterocongrinae | 异康吉鳗属Heteroconger | 哈氏异康吉鳗Heteroconger hassi | AP010859 |
| 康吉鳗亚科Congrinae | 康吉鳗属Conger | 暗康吉鳗Conger erebennus | OM691699 |
| 星康吉鳗Conger myriaster | AB038381 | ||
| 美洲康吉鳗Conger oceanicus | OR546244 | ||
| 日本康吉鳗Conger japonicus | KR131863 | ||
| 异唇鳗属Xenomystax | 似康吉异唇鳗Xenomystax congroides | OR482507 | |
| 异唇鳗Xenomystax atrarius | OR482465 | ||
| 吻鳗属Rhynchoconger | 黑尾吻鳗Rhynchoconger ectenurus | MN256490 | |
| 尖尾鳗属Uroconger | 尖尾鳗Uroconger lepturus | OR581023 | |
| 丝尾鳗Uroconger syringinus | OP056976 | ||
| 深海尾鳗属Bathyuroconger | 深海尾鳗Bathyuroconger vicinus | OP035192 | |
| 深海尾鳗Bathyuroconger vicinus | 本研究 |
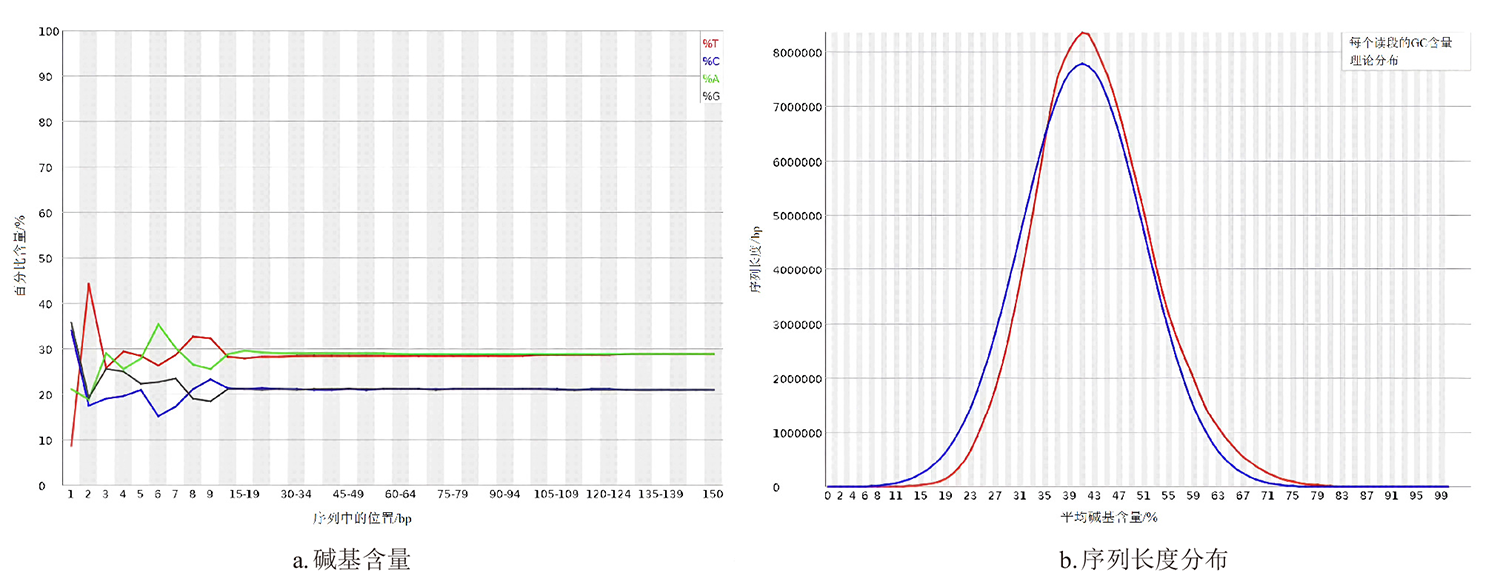
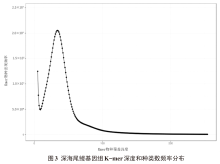
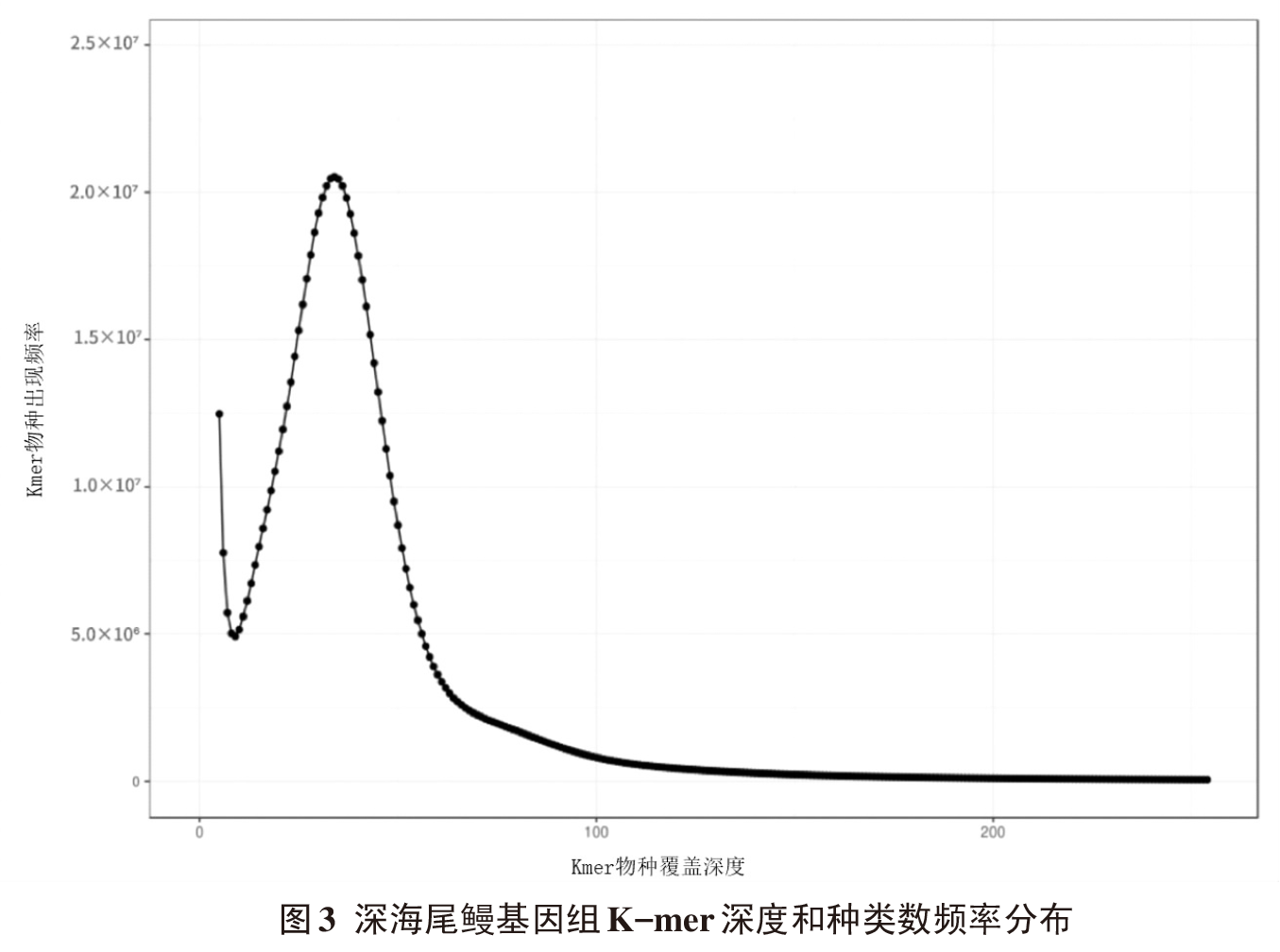
| 组装级别 | 总长度/bp | 序列数量 | 序列数量(≥2 kb) | N50值/bp | GC含量/% |
|---|---|---|---|---|---|
| Contig | 1895343751 | 12514712 | 42840 | 238 | 43.1 |
| Scaffold | 1424281181 | 3643303 | 104841 | 828 | 40.6 |
| 组装级别 | 总长度/bp | 序列数量 | 序列数量(≥2 kb) | N50值/bp | GC含量/% |
|---|---|---|---|---|---|
| Contig | 1895343751 | 12514712 | 42840 | 238 | 43.1 |
| Scaffold | 1424281181 | 3643303 | 104841 | 828 | 40.6 |
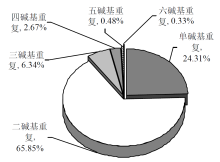
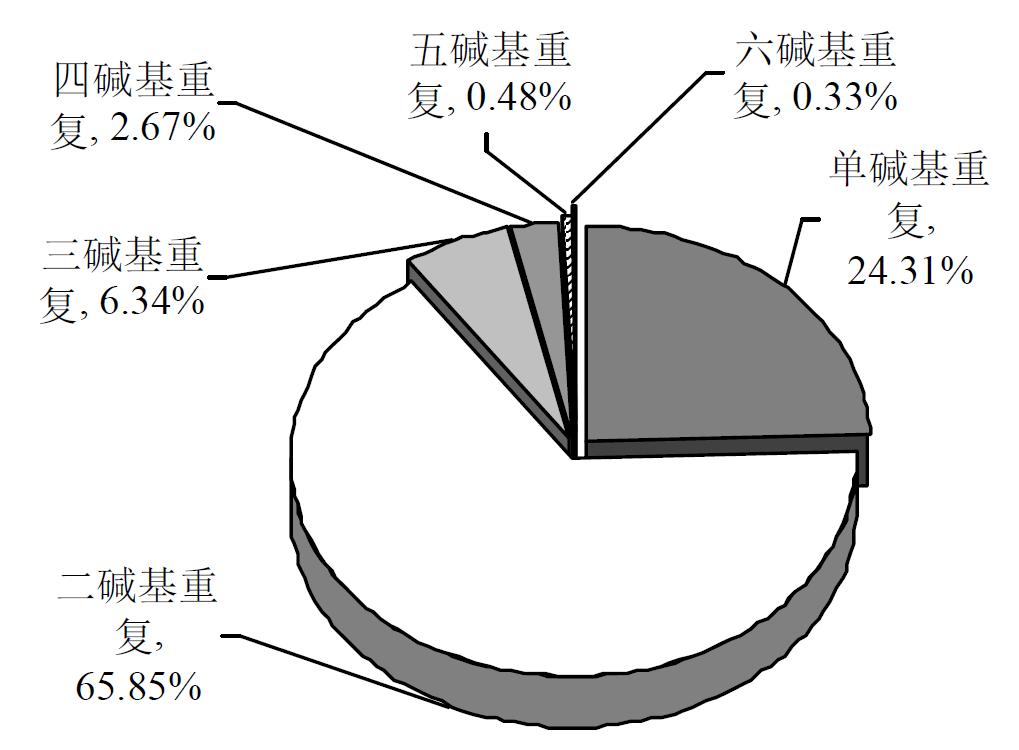
| 重复类型 | 数量 | 出现频率/% | 相对丰度/(个/Mb) | 平均长度/bp | 总长度/bp |
|---|---|---|---|---|---|
| 单碱基重复 | 296884 | 8.15 | 208.44 | 10.73 | 3186246 |
| 二碱基重复 | 804493 | 22.08 | 564.84 | 7.25 | 5835940 |
| 三碱基重复 | 77485 | 2.13 | 54.40 | 14.61 | 1132785 |
| 四碱基重复 | 32633 | 0.90 | 22.91 | 17.36 | 566572 |
| 五碱基重复 | 5852 | 0.16 | 4.11 | 23.21 | 135805 |
| 六碱基重复 | 4087 | 0.11 | 2.87 | 19.45 | 79500 |
| 总计 | 1221434 | 33.53 | 857.57 | 92.61 | 10936848 |
| 重复类型 | 数量 | 出现频率/% | 相对丰度/(个/Mb) | 平均长度/bp | 总长度/bp |
|---|---|---|---|---|---|
| 单碱基重复 | 296884 | 8.15 | 208.44 | 10.73 | 3186246 |
| 二碱基重复 | 804493 | 22.08 | 564.84 | 7.25 | 5835940 |
| 三碱基重复 | 77485 | 2.13 | 54.40 | 14.61 | 1132785 |
| 四碱基重复 | 32633 | 0.90 | 22.91 | 17.36 | 566572 |
| 五碱基重复 | 5852 | 0.16 | 4.11 | 23.21 | 135805 |
| 六碱基重复 | 4087 | 0.11 | 2.87 | 19.45 | 79500 |
| 总计 | 1221434 | 33.53 | 857.57 | 92.61 | 10936848 |
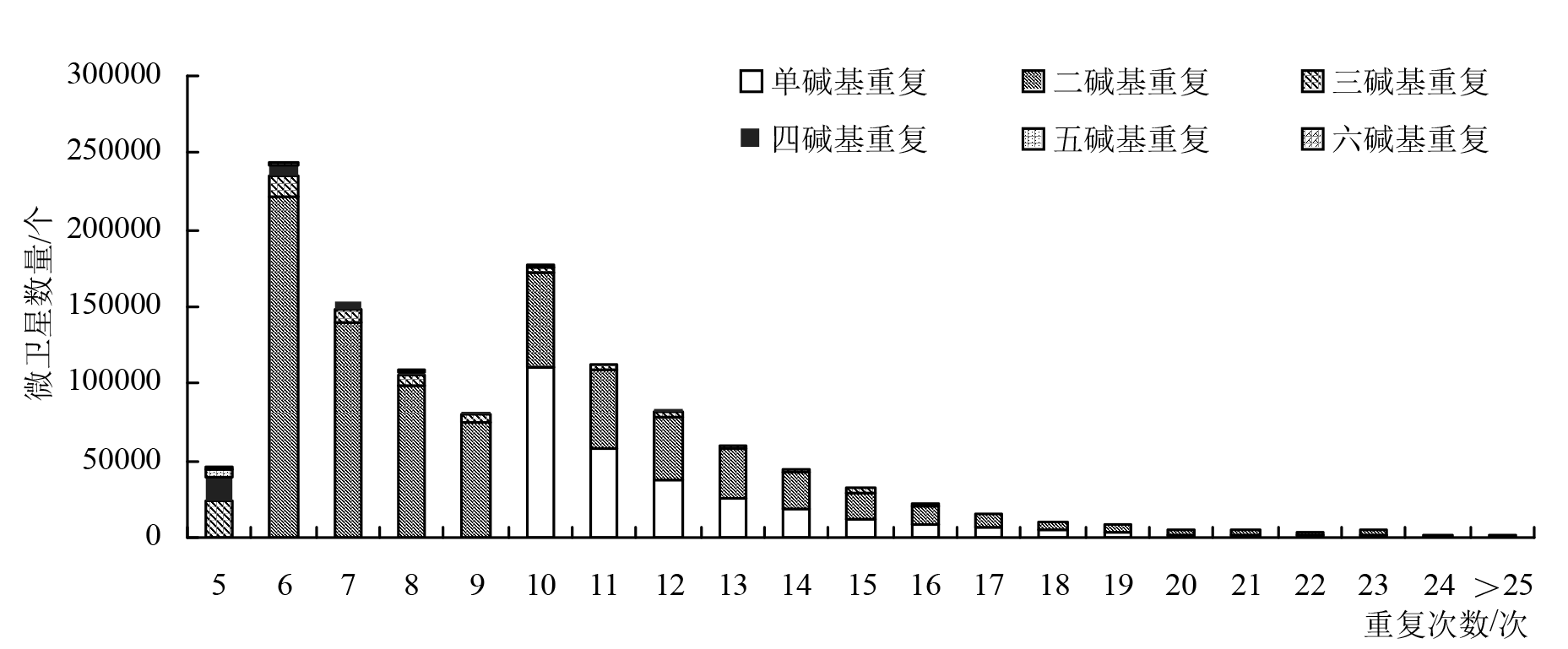
| 重复次数 | 单碱基重复/个 | 二碱基重复/个 | 三碱基重复/个 | 四碱基重复/个 | 五碱基重复/个 | 六碱基重复/个 | 合计/个 | 占比/% |
|---|---|---|---|---|---|---|---|---|
| 5 | 0 | 0 | 24332 | 15525 | 3615 | 1746 | 45218 | 3.70 |
| 6 | 0 | 221468 | 13981 | 7176 | 1041 | 851 | 244517 | 20.02 |
| 7 | 0 | 139104 | 9972 | 3520 | 496 | 595 | 153687 | 12.58 |
| 8 | 0 | 98297 | 7153 | 1851 | 280 | 889 | 108470 | 8.88 |
| 9 | 0 | 75633 | 5270 | 1179 | 200 | 6 | 82288 | 6.74 |
| 10 | 110388 | 61055 | 4006 | 908 | 220 | 0 | 176577 | 14.46 |
| 11 | 58531 | 50354 | 2920 | 890 | 0 | 0 | 112695 | 9.23 |
| 12 | 37803 | 41211 | 2295 | 1472 | 0 | 0 | 82781 | 6.78 |
| 13 | 25846 | 31658 | 1881 | 112 | 0 | 0 | 59497 | 4.87 |
| 14 | 18068 | 23904 | 1614 | 0 | 0 | 0 | 43586 | 3.57 |
| 15 | 12542 | 17012 | 2355 | 0 | 0 | 0 | 31909 | 2.61 |
| 16 | 9291 | 11955 | 1629 | 0 | 0 | 0 | 22875 | 1.87 |
| 17 | 6651 | 8163 | 77 | 0 | 0 | 0 | 14891 | 1.22 |
| 18 | 4814 | 5869 | 0 | 0 | 0 | 0 | 10683 | 0.87 |
| 19 | 3501 | 4235 | 0 | 0 | 0 | 0 | 7736 | 0.63 |
| 20 | 2384 | 3340 | 0 | 0 | 0 | 0 | 5724 | 0.47 |
| 21 | 1807 | 2825 | 0 | 0 | 0 | 0 | 4632 | 0.38 |
| 22 | 1313 | 2852 | 0 | 0 | 0 | 0 | 4165 | 0.34 |
| 23 | 897 | 3860 | 0 | 0 | 0 | 0 | 4757 | 0.39 |
| 24 | 634 | 1635 | 0 | 0 | 0 | 0 | 2269 | 0.19 |
| >25 | 2414 | 63 | 0 | 0 | 0 | 0 | 2477 | 0.20 |
| 重复次数 | 单碱基重复/个 | 二碱基重复/个 | 三碱基重复/个 | 四碱基重复/个 | 五碱基重复/个 | 六碱基重复/个 | 合计/个 | 占比/% |
|---|---|---|---|---|---|---|---|---|
| 5 | 0 | 0 | 24332 | 15525 | 3615 | 1746 | 45218 | 3.70 |
| 6 | 0 | 221468 | 13981 | 7176 | 1041 | 851 | 244517 | 20.02 |
| 7 | 0 | 139104 | 9972 | 3520 | 496 | 595 | 153687 | 12.58 |
| 8 | 0 | 98297 | 7153 | 1851 | 280 | 889 | 108470 | 8.88 |
| 9 | 0 | 75633 | 5270 | 1179 | 200 | 6 | 82288 | 6.74 |
| 10 | 110388 | 61055 | 4006 | 908 | 220 | 0 | 176577 | 14.46 |
| 11 | 58531 | 50354 | 2920 | 890 | 0 | 0 | 112695 | 9.23 |
| 12 | 37803 | 41211 | 2295 | 1472 | 0 | 0 | 82781 | 6.78 |
| 13 | 25846 | 31658 | 1881 | 112 | 0 | 0 | 59497 | 4.87 |
| 14 | 18068 | 23904 | 1614 | 0 | 0 | 0 | 43586 | 3.57 |
| 15 | 12542 | 17012 | 2355 | 0 | 0 | 0 | 31909 | 2.61 |
| 16 | 9291 | 11955 | 1629 | 0 | 0 | 0 | 22875 | 1.87 |
| 17 | 6651 | 8163 | 77 | 0 | 0 | 0 | 14891 | 1.22 |
| 18 | 4814 | 5869 | 0 | 0 | 0 | 0 | 10683 | 0.87 |
| 19 | 3501 | 4235 | 0 | 0 | 0 | 0 | 7736 | 0.63 |
| 20 | 2384 | 3340 | 0 | 0 | 0 | 0 | 5724 | 0.47 |
| 21 | 1807 | 2825 | 0 | 0 | 0 | 0 | 4632 | 0.38 |
| 22 | 1313 | 2852 | 0 | 0 | 0 | 0 | 4165 | 0.34 |
| 23 | 897 | 3860 | 0 | 0 | 0 | 0 | 4757 | 0.39 |
| 24 | 634 | 1635 | 0 | 0 | 0 | 0 | 2269 | 0.19 |
| >25 | 2414 | 63 | 0 | 0 | 0 | 0 | 2477 | 0.20 |

| 重复类型 | 种类/种 | 最多重复基元 | 最少重复基元 | |||||
|---|---|---|---|---|---|---|---|---|
| 重复基元 | 数量/个 | 占比/% | 重复基元 | 数量/个 | 占比/% | |||
| 单碱基重复 | 4 | A | 128843 | 43.40 | G | 29248 | 9.85 | |
| 二碱基重复 | 12 | CA | 239966 | 29.83 | GC | 522 | 0.06 | |
| 三碱基重复 | 60 | AAT | 9533 | 12.30 | ACG | 14 | 0.02 | |
| 四碱基重复 | 231 | AAAT | 2663 | 8.16 | ACGA/ACGT/ATCG/ CCCA/CCGA/CGAA/ CGAT/CGTA/TACG/TCGG | 1 | 0.00 | |
| 五碱基重复 | 583 | CATTA | 316 | 5.40 | — | 1 | 0.02 | |
| 六碱基重复 | 668 | CACACT | 268 | 6.56 | — | 1 | 0.02 | |
| 重复类型 | 种类/种 | 最多重复基元 | 最少重复基元 | |||||
|---|---|---|---|---|---|---|---|---|
| 重复基元 | 数量/个 | 占比/% | 重复基元 | 数量/个 | 占比/% | |||
| 单碱基重复 | 4 | A | 128843 | 43.40 | G | 29248 | 9.85 | |
| 二碱基重复 | 12 | CA | 239966 | 29.83 | GC | 522 | 0.06 | |
| 三碱基重复 | 60 | AAT | 9533 | 12.30 | ACG | 14 | 0.02 | |
| 四碱基重复 | 231 | AAAT | 2663 | 8.16 | ACGA/ACGT/ATCG/ CCCA/CCGA/CGAA/ CGAT/CGTA/TACG/TCGG | 1 | 0.00 | |
| 五碱基重复 | 583 | CATTA | 316 | 5.40 | — | 1 | 0.02 | |
| 六碱基重复 | 668 | CACACT | 268 | 6.56 | — | 1 | 0.02 | |
| [1] |
陈大刚, 张美昭. 中国海洋鱼类[M]. 青岛: 中国海洋大学出版社, 2015.
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
高胜寒, 禹海英, 吴双阳, 等. 复杂基因组测序技术研究进展[J]. 遗传, 2018, 40(11):944-963.
|
| [7] |
|
| [8] |
|
| [9] |
朱滨, 常剑波. 微卫星DNA及其在鱼类中的应用[J]. 水生生物学报, 1999, 23(6):721-729.
|
| [10] |
孙效文, 张晓锋, 赵莹莹, 等. 水产生物微卫星标记技术研究进展及其应用[J]. 中国水产科学, 2008, 15(4):689-703.
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
doi: 10.1093/bioinformatics/bty560 pmid: 30423086 |
| [15] |
|
| [16] |
doi: 10.1186/2047-217X-1-18 pmid: 23587118 |
| [17] |
|
| [18] |
doi: 10.1093/bioinformatics/btx198 pmid: 28398459 |
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
胡震, 曹俊. 载人深潜技术的发展与应用[J]. 中国工程科学, 2019, 21(6):87-96.
doi: 10.15302/J-SSCAE-2019.06.017 |
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
doi: 10.1038/s41597-024-03817-9 pmid: 39214993 |
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
pmid: 16213058 |
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
doi: 10.1093/bioinformatics/btr011 pmid: 21217122 |
| [40] |
刘焘, 陈冰洁, 史会来, 等. 基于基因组survey的横带髭鲷(Hapalogenys analis)微卫星位点筛选与特征分析[J]. 海洋与湖沼, 2023, 54(3):848-855.
|
| [41] |
刘焘. 三种海洋鱼类全基因组Survey分析及SSR位点挖掘[D]. 舟山: 浙江海洋大学, 2023.
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
doi: 10.1093/nar/20.2.211 pmid: 1741246 |
| [46] |
doi: 10.1093/genetics/146.3.769 pmid: 9215886 |
| [47] |
黄新芯, 蒋艳琳, 蒋小姿, 等. 基于高通量转录组测序技术的龙头鱼微卫星信息分析[J]. 浙江海洋大学学报(自然科学版), 2021, 40(3):189-197.
|
| [48] |
刘玉萍, 王棋, 黄新芯, 等. 基于高通量测序的带鱼肌肉组织转录组微卫星信息分析[J]. 南方农业学报, 2022, 53(3):725-734.
|
| [49] |
|
| [50] |
|
| [51] |
|
| [1] | 王长秘, 罗志明, 李银煳, 王晓燕, 张荣跃, 李婕, 尹炯, 单红丽. 甘蔗梢腐病致病菌甘蔗镰刀菌的分离及鉴定[J]. 中国农学通报, 2025, 41(4): 114-118. |
| [2] | 韩垚, 杨小艳, 谢树章. 植物组培中污染微生物的分离鉴定及药剂筛选[J]. 中国农学通报, 2025, 41(3): 131-138. |
| [3] | 饶永斌, 张君丽. 一株野生肉色隔孢伏革菌的分子鉴定、生物学特性及驯化[J]. 中国农学通报, 2025, 41(3): 123-130. |
| [4] | 康美花, 段灵涛, 阴长发, 曲润波, 肖苏军, 王希, 张露, 况虹敏, 孙强, 陈洪凡, 杨迎青, 邵见阳, 涂雪琴, 兰波. 水稻立枯病菌的鉴定及其系统进化分析[J]. 中国农学通报, 2024, 40(36): 140-146. |
| [5] | 王越, 曹春梅, 陈汉, 王晓娇, 余乾鹏, 李学洋, 张志凯, 胡柏耿. 乌兰察布地区马铃薯疮痂病新型病原菌种类及致病性鉴定[J]. 中国农学通报, 2024, 40(32): 151-156. |
| [6] | 高凯利, 马学艳, 王海华, 李涵, 李燕华, 金武, 闻海波. 5个宽体金线蛭种群遗传多样性和遗传分化的SNP分析[J]. 中国农学通报, 2024, 40(29): 144-149. |
| [7] | 孙保娟, 李涛, 游倩, 宫超, 黎振兴, 李植良. 茄科植物花青素合成相关的MYB转录因子研究进展[J]. 中国农学通报, 2023, 39(36): 102-111. |
| [8] | 努尔夏提·努尔买买提, 李焕宇, 杨成德. 辣椒根腐病菌拮抗链霉菌的筛选及16S rRNA基因系统发育分析[J]. 中国农学通报, 2022, 38(33): 116-123. |
| [9] | 高忠奎, 蒋菁, 韩柱强, 黄志鹏, 熊发前, 唐秀梅, 吴海宁, 钟瑞春, 刘菁, 唐荣华, 贺梁琼. CRISPR/Cas9系统及其在粮油作物遗传改良中的研究进展[J]. 中国农学通报, 2021, 37(20): 26-34. |
| [10] | 高杜娟, 刘兴录, 兰志斌, 赵杨, 陈友德, 周斌, 吕艳梅, 罗先富, 唐善军. 水稻-油菜周年耕作方式对土壤微生态的影响[J]. 中国农学通报, 2021, 37(16): 74-81. |
| [11] | 孙雨茜, 倪万潮, 杜平, 崔晓霞, 郭书巧, 蒋璐. 基于简化基因组的甜叶菊分子标记开发[J]. 中国农学通报, 2020, 36(27): 111-117. |
| [12] | 周梦晨, 徐东坡, 方弟安, 周彦锋, 刘凯, 张敏莹. 6个不同群体鳜基于SSR的遗传多样性初步研究[J]. 中国农学通报, 2020, 36(26): 147-152. |
| [13] | 程志强,雷少楠,熊娟,马荣琴,陈优优,李容丹,路晶晶,吴寒,龚玉杰,田宝玉. 番茄根内生芽孢杆菌的多样性和系统发育研究[J]. 中国农学通报, 2018, 34(8): 37-45. |
| [14] | 卢钰升,顾文杰,蒋瑞萍,孙丽丽,徐培智,谭志远,解开治,李文英,李 夏. 一株生防细菌GB58的鉴定与抑菌能力测定[J]. 中国农学通报, 2016, 32(6): 198-204. |
| [15] | 朱 玲,徐崇志,张 锐. 新疆扁桃种质资源ITS序列分析及系统发育研究[J]. 中国农学通报, 2016, 32(4): 155-159. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||