Welcome to Chinese Agricultural Science Bulletin,
Chinese Agricultural Science Bulletin ›› 2025, Vol. 41 ›› Issue (25): 153-164.doi: 10.11924/j.issn.1000-6850.casb2025-0057
ZHENG Yunkai( ), LIU Penghui, NIU Shiyang, YANG Tianyan()
), LIU Penghui, NIU Shiyang, YANG Tianyan()
Received:2025-01-16
Revised:2025-06-03
Online:2025-09-05
Published:2025-09-16
ZHENG Yunkai, LIU Penghui, NIU Shiyang, YANG Tianyan. Analysis of Microsatellites Characterization and Phylogenetic Relationship for Bathyuroconger vicinus Based on Genomic Survey Data[J]. Chinese Agricultural Science Bulletin, 2025, 41(25): 153-164.
Add to citation manager EndNote|Ris|BibTeX
URL: https://www.casb.org.cn/EN/10.11924/j.issn.1000-6850.casb2025-0057
| 亚科 | 属 | 种 | GenBank序列号 |
|---|---|---|---|
| 深海康吉鳗亚科Bathymyrinae | 美体鳗属Ariosoma | 梅氏美体鳗Ariosoma meeki | OK585090 |
| 细美体鳗Ariosoma shiroanago | AP010861 | ||
| 副康吉鳗属Paraconger | 几内亚副康吉鳗Paraconger notialis | AP010860 | |
| 尾边副康吉鳗Paraconger caudilimbatus | OR582666 | ||
| 异康吉鳗亚科Heterocongrinae | 异康吉鳗属Heteroconger | 哈氏异康吉鳗Heteroconger hassi | AP010859 |
| 康吉鳗亚科Congrinae | 康吉鳗属Conger | 暗康吉鳗Conger erebennus | OM691699 |
| 星康吉鳗Conger myriaster | AB038381 | ||
| 美洲康吉鳗Conger oceanicus | OR546244 | ||
| 日本康吉鳗Conger japonicus | KR131863 | ||
| 异唇鳗属Xenomystax | 似康吉异唇鳗Xenomystax congroides | OR482507 | |
| 异唇鳗Xenomystax atrarius | OR482465 | ||
| 吻鳗属Rhynchoconger | 黑尾吻鳗Rhynchoconger ectenurus | MN256490 | |
| 尖尾鳗属Uroconger | 尖尾鳗Uroconger lepturus | OR581023 | |
| 丝尾鳗Uroconger syringinus | OP056976 | ||
| 深海尾鳗属Bathyuroconger | 深海尾鳗Bathyuroconger vicinus | OP035192 | |
| 深海尾鳗Bathyuroconger vicinus | 本研究 |
| 亚科 | 属 | 种 | GenBank序列号 |
|---|---|---|---|
| 深海康吉鳗亚科Bathymyrinae | 美体鳗属Ariosoma | 梅氏美体鳗Ariosoma meeki | OK585090 |
| 细美体鳗Ariosoma shiroanago | AP010861 | ||
| 副康吉鳗属Paraconger | 几内亚副康吉鳗Paraconger notialis | AP010860 | |
| 尾边副康吉鳗Paraconger caudilimbatus | OR582666 | ||
| 异康吉鳗亚科Heterocongrinae | 异康吉鳗属Heteroconger | 哈氏异康吉鳗Heteroconger hassi | AP010859 |
| 康吉鳗亚科Congrinae | 康吉鳗属Conger | 暗康吉鳗Conger erebennus | OM691699 |
| 星康吉鳗Conger myriaster | AB038381 | ||
| 美洲康吉鳗Conger oceanicus | OR546244 | ||
| 日本康吉鳗Conger japonicus | KR131863 | ||
| 异唇鳗属Xenomystax | 似康吉异唇鳗Xenomystax congroides | OR482507 | |
| 异唇鳗Xenomystax atrarius | OR482465 | ||
| 吻鳗属Rhynchoconger | 黑尾吻鳗Rhynchoconger ectenurus | MN256490 | |
| 尖尾鳗属Uroconger | 尖尾鳗Uroconger lepturus | OR581023 | |
| 丝尾鳗Uroconger syringinus | OP056976 | ||
| 深海尾鳗属Bathyuroconger | 深海尾鳗Bathyuroconger vicinus | OP035192 | |
| 深海尾鳗Bathyuroconger vicinus | 本研究 |
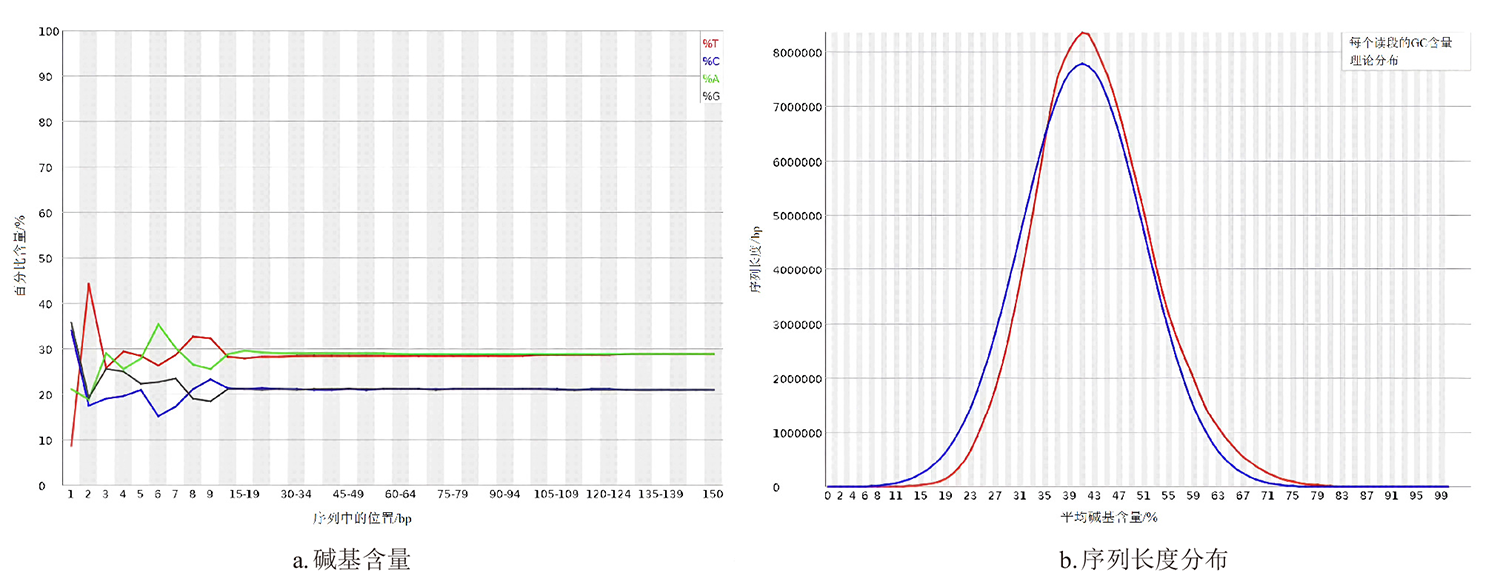
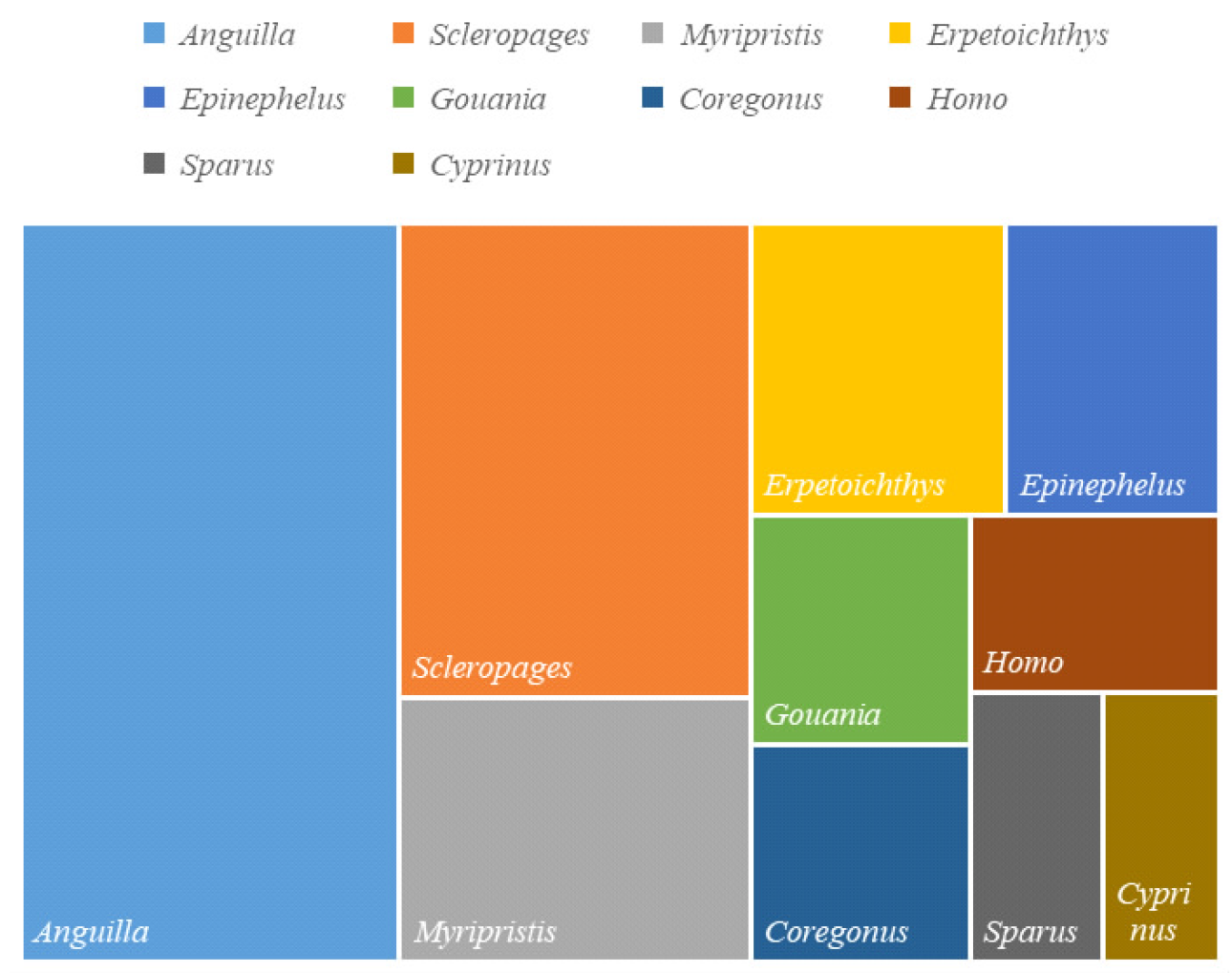
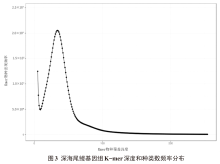
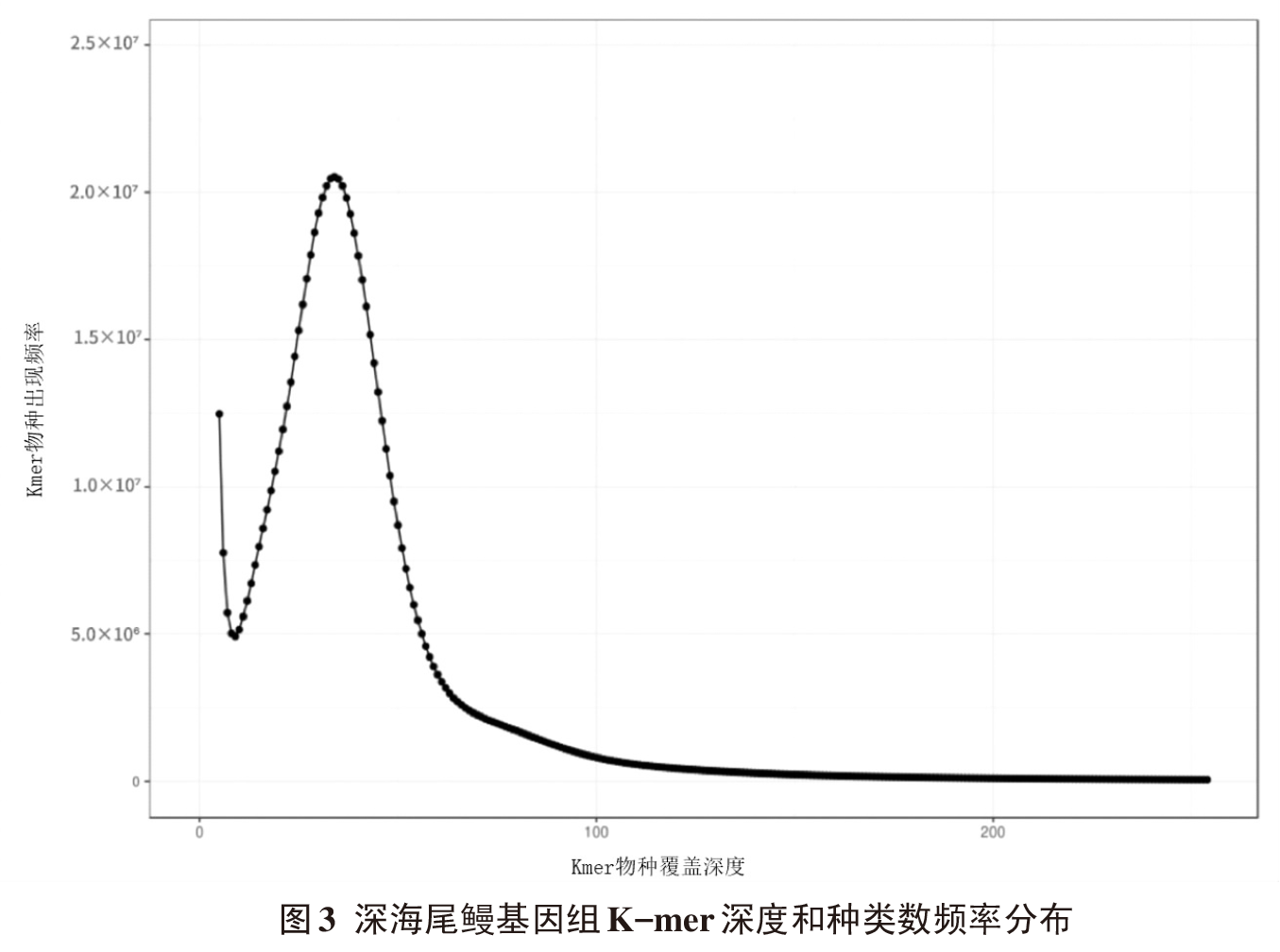
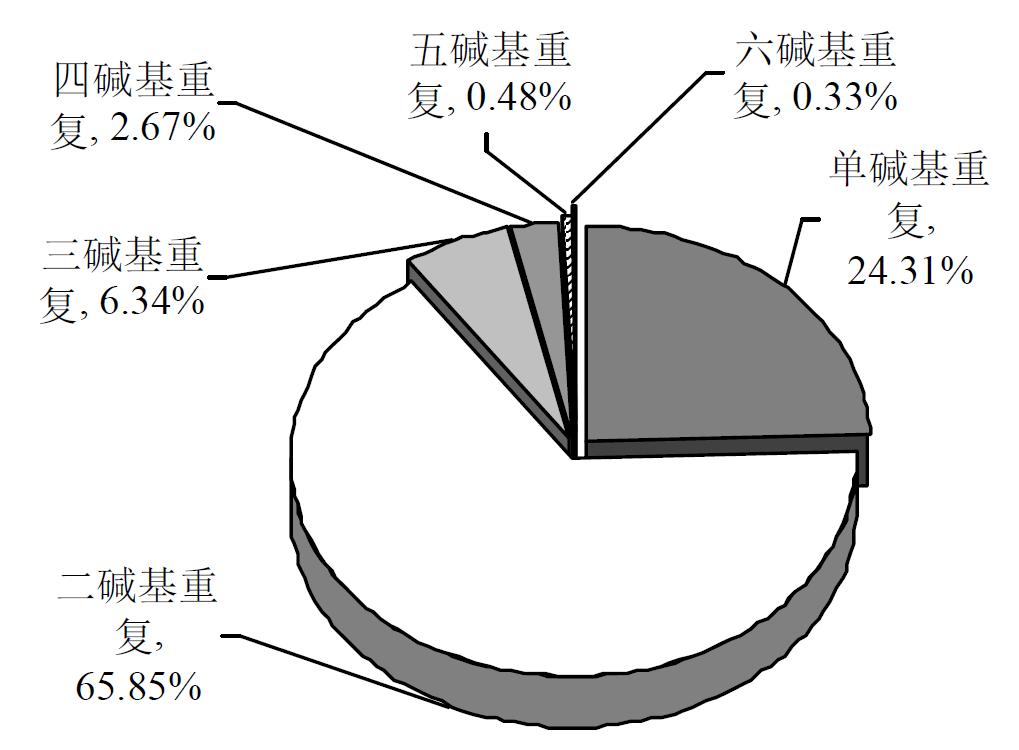
| 重复类型 | 数量 | 出现频率/% | 相对丰度/(个/Mb) | 平均长度/bp | 总长度/bp |
|---|---|---|---|---|---|
| 单碱基重复 | 296884 | 8.15 | 208.44 | 10.73 | 3186246 |
| 二碱基重复 | 804493 | 22.08 | 564.84 | 7.25 | 5835940 |
| 三碱基重复 | 77485 | 2.13 | 54.40 | 14.61 | 1132785 |
| 四碱基重复 | 32633 | 0.90 | 22.91 | 17.36 | 566572 |
| 五碱基重复 | 5852 | 0.16 | 4.11 | 23.21 | 135805 |
| 六碱基重复 | 4087 | 0.11 | 2.87 | 19.45 | 79500 |
| 总计 | 1221434 | 33.53 | 857.57 | 92.61 | 10936848 |
| 重复类型 | 数量 | 出现频率/% | 相对丰度/(个/Mb) | 平均长度/bp | 总长度/bp |
|---|---|---|---|---|---|
| 单碱基重复 | 296884 | 8.15 | 208.44 | 10.73 | 3186246 |
| 二碱基重复 | 804493 | 22.08 | 564.84 | 7.25 | 5835940 |
| 三碱基重复 | 77485 | 2.13 | 54.40 | 14.61 | 1132785 |
| 四碱基重复 | 32633 | 0.90 | 22.91 | 17.36 | 566572 |
| 五碱基重复 | 5852 | 0.16 | 4.11 | 23.21 | 135805 |
| 六碱基重复 | 4087 | 0.11 | 2.87 | 19.45 | 79500 |
| 总计 | 1221434 | 33.53 | 857.57 | 92.61 | 10936848 |
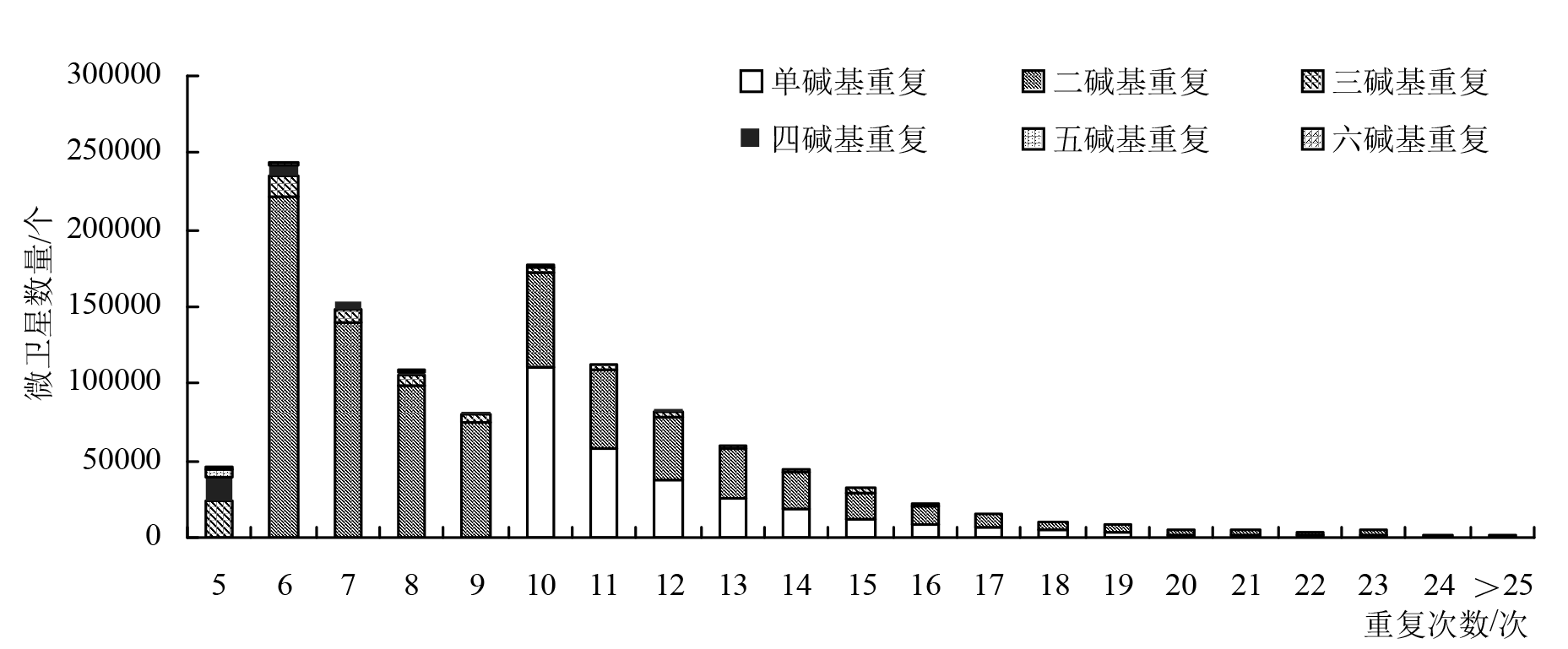
| 重复次数 | 单碱基重复/个 | 二碱基重复/个 | 三碱基重复/个 | 四碱基重复/个 | 五碱基重复/个 | 六碱基重复/个 | 合计/个 | 占比/% |
|---|---|---|---|---|---|---|---|---|
| 5 | 0 | 0 | 24332 | 15525 | 3615 | 1746 | 45218 | 3.70 |
| 6 | 0 | 221468 | 13981 | 7176 | 1041 | 851 | 244517 | 20.02 |
| 7 | 0 | 139104 | 9972 | 3520 | 496 | 595 | 153687 | 12.58 |
| 8 | 0 | 98297 | 7153 | 1851 | 280 | 889 | 108470 | 8.88 |
| 9 | 0 | 75633 | 5270 | 1179 | 200 | 6 | 82288 | 6.74 |
| 10 | 110388 | 61055 | 4006 | 908 | 220 | 0 | 176577 | 14.46 |
| 11 | 58531 | 50354 | 2920 | 890 | 0 | 0 | 112695 | 9.23 |
| 12 | 37803 | 41211 | 2295 | 1472 | 0 | 0 | 82781 | 6.78 |
| 13 | 25846 | 31658 | 1881 | 112 | 0 | 0 | 59497 | 4.87 |
| 14 | 18068 | 23904 | 1614 | 0 | 0 | 0 | 43586 | 3.57 |
| 15 | 12542 | 17012 | 2355 | 0 | 0 | 0 | 31909 | 2.61 |
| 16 | 9291 | 11955 | 1629 | 0 | 0 | 0 | 22875 | 1.87 |
| 17 | 6651 | 8163 | 77 | 0 | 0 | 0 | 14891 | 1.22 |
| 18 | 4814 | 5869 | 0 | 0 | 0 | 0 | 10683 | 0.87 |
| 19 | 3501 | 4235 | 0 | 0 | 0 | 0 | 7736 | 0.63 |
| 20 | 2384 | 3340 | 0 | 0 | 0 | 0 | 5724 | 0.47 |
| 21 | 1807 | 2825 | 0 | 0 | 0 | 0 | 4632 | 0.38 |
| 22 | 1313 | 2852 | 0 | 0 | 0 | 0 | 4165 | 0.34 |
| 23 | 897 | 3860 | 0 | 0 | 0 | 0 | 4757 | 0.39 |
| 24 | 634 | 1635 | 0 | 0 | 0 | 0 | 2269 | 0.19 |
| >25 | 2414 | 63 | 0 | 0 | 0 | 0 | 2477 | 0.20 |
| 重复次数 | 单碱基重复/个 | 二碱基重复/个 | 三碱基重复/个 | 四碱基重复/个 | 五碱基重复/个 | 六碱基重复/个 | 合计/个 | 占比/% |
|---|---|---|---|---|---|---|---|---|
| 5 | 0 | 0 | 24332 | 15525 | 3615 | 1746 | 45218 | 3.70 |
| 6 | 0 | 221468 | 13981 | 7176 | 1041 | 851 | 244517 | 20.02 |
| 7 | 0 | 139104 | 9972 | 3520 | 496 | 595 | 153687 | 12.58 |
| 8 | 0 | 98297 | 7153 | 1851 | 280 | 889 | 108470 | 8.88 |
| 9 | 0 | 75633 | 5270 | 1179 | 200 | 6 | 82288 | 6.74 |
| 10 | 110388 | 61055 | 4006 | 908 | 220 | 0 | 176577 | 14.46 |
| 11 | 58531 | 50354 | 2920 | 890 | 0 | 0 | 112695 | 9.23 |
| 12 | 37803 | 41211 | 2295 | 1472 | 0 | 0 | 82781 | 6.78 |
| 13 | 25846 | 31658 | 1881 | 112 | 0 | 0 | 59497 | 4.87 |
| 14 | 18068 | 23904 | 1614 | 0 | 0 | 0 | 43586 | 3.57 |
| 15 | 12542 | 17012 | 2355 | 0 | 0 | 0 | 31909 | 2.61 |
| 16 | 9291 | 11955 | 1629 | 0 | 0 | 0 | 22875 | 1.87 |
| 17 | 6651 | 8163 | 77 | 0 | 0 | 0 | 14891 | 1.22 |
| 18 | 4814 | 5869 | 0 | 0 | 0 | 0 | 10683 | 0.87 |
| 19 | 3501 | 4235 | 0 | 0 | 0 | 0 | 7736 | 0.63 |
| 20 | 2384 | 3340 | 0 | 0 | 0 | 0 | 5724 | 0.47 |
| 21 | 1807 | 2825 | 0 | 0 | 0 | 0 | 4632 | 0.38 |
| 22 | 1313 | 2852 | 0 | 0 | 0 | 0 | 4165 | 0.34 |
| 23 | 897 | 3860 | 0 | 0 | 0 | 0 | 4757 | 0.39 |
| 24 | 634 | 1635 | 0 | 0 | 0 | 0 | 2269 | 0.19 |
| >25 | 2414 | 63 | 0 | 0 | 0 | 0 | 2477 | 0.20 |

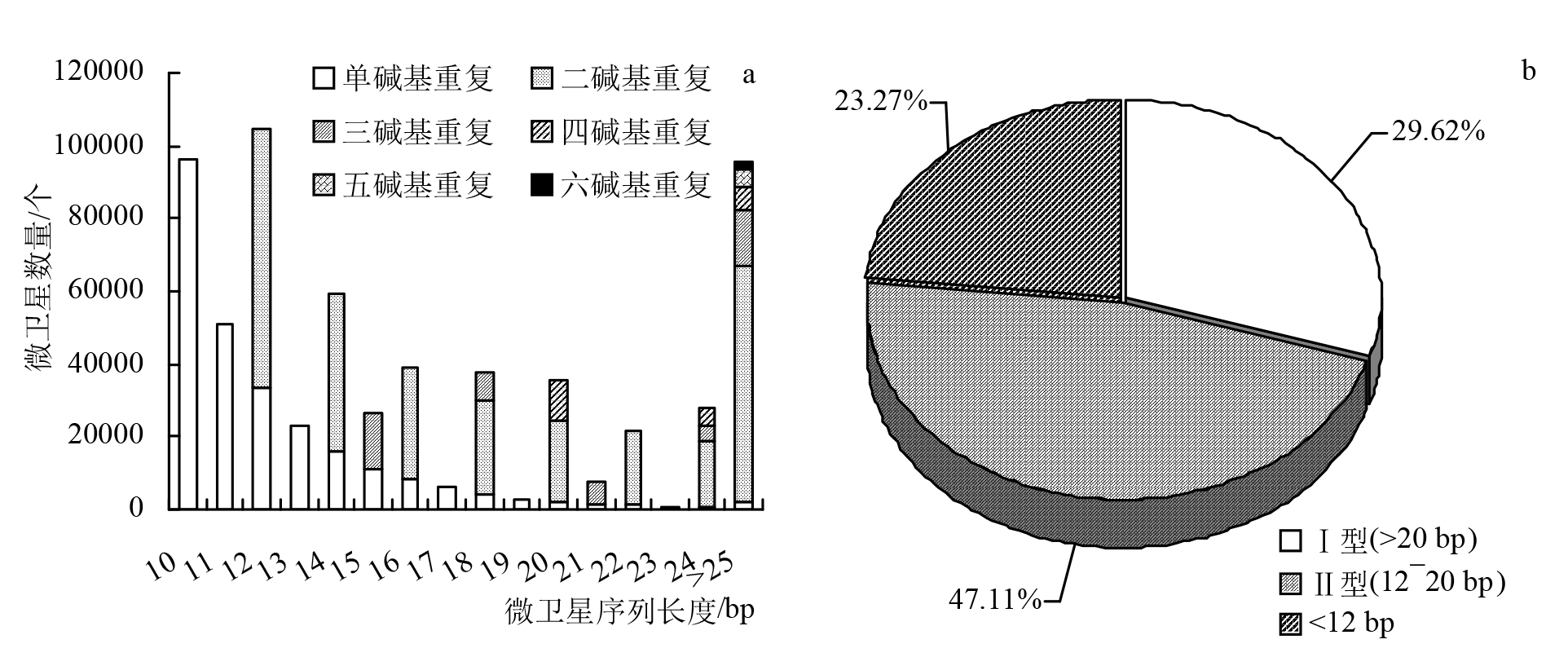
| 重复类型 | 种类/种 | 最多重复基元 | 最少重复基元 | |||||
|---|---|---|---|---|---|---|---|---|
| 重复基元 | 数量/个 | 占比/% | 重复基元 | 数量/个 | 占比/% | |||
| 单碱基重复 | 4 | A | 128843 | 43.40 | G | 29248 | 9.85 | |
| 二碱基重复 | 12 | CA | 239966 | 29.83 | GC | 522 | 0.06 | |
| 三碱基重复 | 60 | AAT | 9533 | 12.30 | ACG | 14 | 0.02 | |
| 四碱基重复 | 231 | AAAT | 2663 | 8.16 | ACGA/ACGT/ATCG/ CCCA/CCGA/CGAA/ CGAT/CGTA/TACG/TCGG | 1 | 0.00 | |
| 五碱基重复 | 583 | CATTA | 316 | 5.40 | — | 1 | 0.02 | |
| 六碱基重复 | 668 | CACACT | 268 | 6.56 | — | 1 | 0.02 | |
| 重复类型 | 种类/种 | 最多重复基元 | 最少重复基元 | |||||
|---|---|---|---|---|---|---|---|---|
| 重复基元 | 数量/个 | 占比/% | 重复基元 | 数量/个 | 占比/% | |||
| 单碱基重复 | 4 | A | 128843 | 43.40 | G | 29248 | 9.85 | |
| 二碱基重复 | 12 | CA | 239966 | 29.83 | GC | 522 | 0.06 | |
| 三碱基重复 | 60 | AAT | 9533 | 12.30 | ACG | 14 | 0.02 | |
| 四碱基重复 | 231 | AAAT | 2663 | 8.16 | ACGA/ACGT/ATCG/ CCCA/CCGA/CGAA/ CGAT/CGTA/TACG/TCGG | 1 | 0.00 | |
| 五碱基重复 | 583 | CATTA | 316 | 5.40 | — | 1 | 0.02 | |
| 六碱基重复 | 668 | CACACT | 268 | 6.56 | — | 1 | 0.02 | |
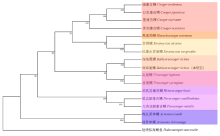
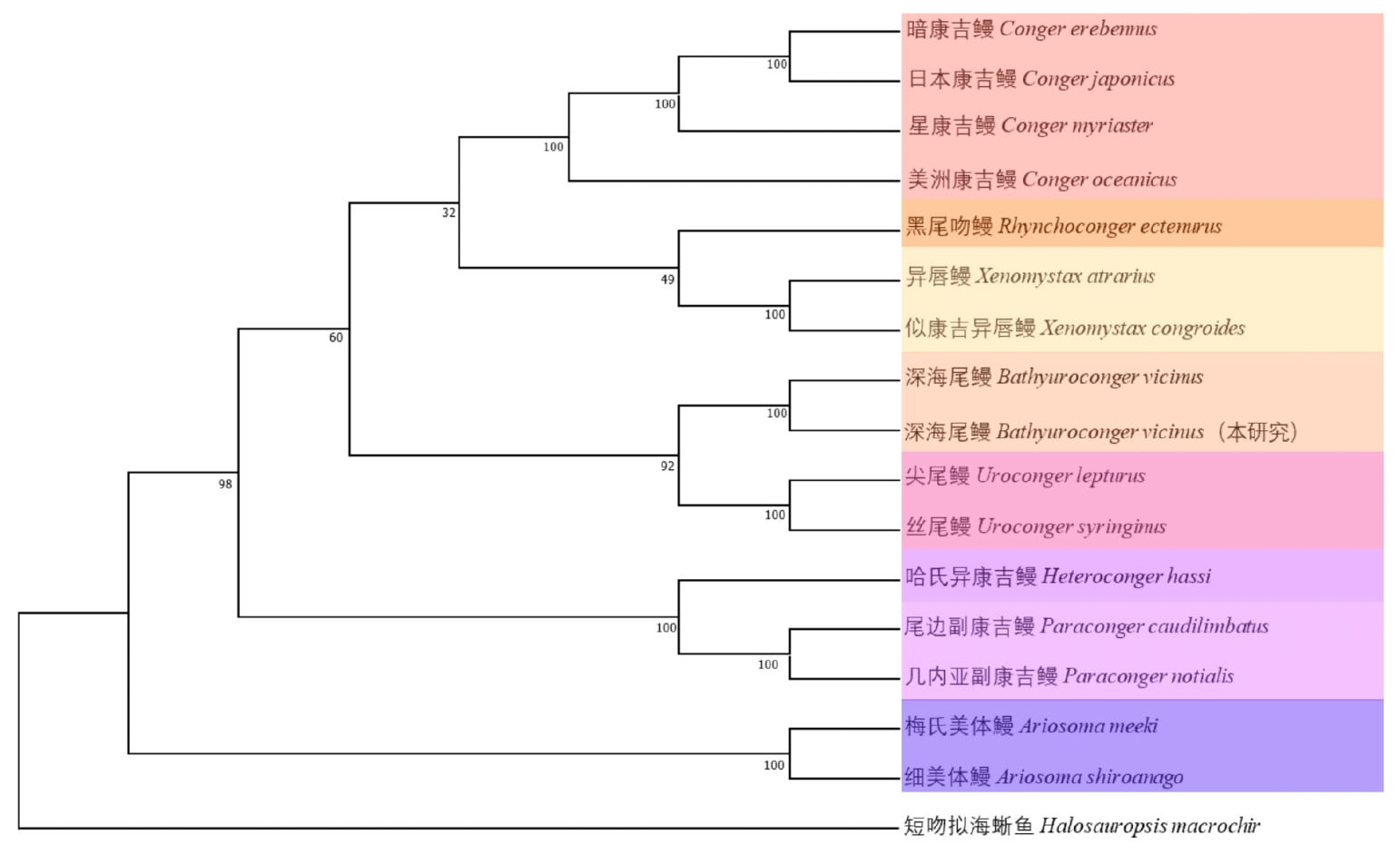
| [1] |
陈大刚, 张美昭. 中国海洋鱼类[M]. 青岛: 中国海洋大学出版社, 2015.
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
高胜寒, 禹海英, 吴双阳, 等. 复杂基因组测序技术研究进展[J]. 遗传, 2018, 40(11):944-963.
|
| [7] |
|
| [8] |
|
| [9] |
朱滨, 常剑波. 微卫星DNA及其在鱼类中的应用[J]. 水生生物学报, 1999, 23(6):721-729.
|
| [10] |
孙效文, 张晓锋, 赵莹莹, 等. 水产生物微卫星标记技术研究进展及其应用[J]. 中国水产科学, 2008, 15(4):689-703.
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
doi: 10.1093/bioinformatics/bty560 pmid: 30423086 |
| [15] |
|
| [16] |
doi: 10.1186/2047-217X-1-18 pmid: 23587118 |
| [17] |
|
| [18] |
doi: 10.1093/bioinformatics/btx198 pmid: 28398459 |
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
胡震, 曹俊. 载人深潜技术的发展与应用[J]. 中国工程科学, 2019, 21(6):87-96.
doi: 10.15302/J-SSCAE-2019.06.017 |
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
doi: 10.1038/s41597-024-03817-9 pmid: 39214993 |
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
pmid: 16213058 |
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
doi: 10.1093/bioinformatics/btr011 pmid: 21217122 |
| [40] |
刘焘, 陈冰洁, 史会来, 等. 基于基因组survey的横带髭鲷(Hapalogenys analis)微卫星位点筛选与特征分析[J]. 海洋与湖沼, 2023, 54(3):848-855.
|
| [41] |
刘焘. 三种海洋鱼类全基因组Survey分析及SSR位点挖掘[D]. 舟山: 浙江海洋大学, 2023.
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
doi: 10.1093/nar/20.2.211 pmid: 1741246 |
| [46] |
doi: 10.1093/genetics/146.3.769 pmid: 9215886 |
| [47] |
黄新芯, 蒋艳琳, 蒋小姿, 等. 基于高通量转录组测序技术的龙头鱼微卫星信息分析[J]. 浙江海洋大学学报(自然科学版), 2021, 40(3):189-197.
|
| [48] |
刘玉萍, 王棋, 黄新芯, 等. 基于高通量测序的带鱼肌肉组织转录组微卫星信息分析[J]. 南方农业学报, 2022, 53(3):725-734.
|
| [49] |
|
| [50] |
|
| [51] |
|
| [1] | WANG Changmi, LUO Zhiming, LI Yinhu, WANG Xiaoyan, ZHANG Rongyue, LI Jie, YIN Jiong, SHAN Hongli. Isolation and Identification of Pathogenic Fungus Fusarium sacchari of Sugarcane Pokkah Boeng Disease [J]. Chinese Agricultural Science Bulletin, 2025, 41(4): 114-118. |
| [2] | HAN Yao, YANG Xiaoyan, XIE Shuzhang. Contaminated Microorganisms in Plant Tissue Culture: Isolation and Identification and Fungicides Screening [J]. Chinese Agricultural Science Bulletin, 2025, 41(3): 131-138. |
| [3] | RAO Yongbin, ZHANG Junli. Molecular Identification, Biological Characteristics and Domestication of A Wild Strain of Peniophora incarnata [J]. Chinese Agricultural Science Bulletin, 2025, 41(3): 123-130. |
| [4] | WANG Yunxuan, LIU Li, SUN Chunhui, LU Shuhao, LIANG Yuqing, CUI Wenli, GAO Jiayi, LI Yongqiang. Ligustrum Virus A Isolates from Syringa lindl: Identification and Whole Genome Sequence Analysis [J]. Chinese Agricultural Science Bulletin, 2025, 41(3): 107-113. |
| [5] | ZHAN Jiatao, MIAO Leyi, JI Zixian, WANG Yiting, LU Yue, TAN En, MA Shuaipeng, WANG Limin. Response Analysis of OsSIG5 Gene to Phytohormones and Abiotic Stresses in Rice [J]. Chinese Agricultural Science Bulletin, 2025, 41(14): 32-40. |
| [6] | KANG Meihua, DUAN Lingtao, YIN Changfa, QU Runbo, XIAO Sujun, WANG Xi, ZHANG Lu, KUANG Hongmin, SUN Qiang, CHEN Hongfan, YANG Yingqing, SHAO Jianyang, TU Xueqin, LAN Bo. Identification and Phylogenetic Analysis of Pathogen of Rice Seedling Blight [J]. Chinese Agricultural Science Bulletin, 2024, 40(36): 140-146. |
| [7] | XU Shiqiang, LI Jingyu, SUN Mingyang, GU Yan, WANG Jihua. Identification and Expression Analysis of NAC Gene Family in Andrographis paniculate [J]. Chinese Agricultural Science Bulletin, 2024, 40(36): 117-125. |
| [8] | WANG Yue, CAO Chunmei, CHEN Han, WANG Xiaojiao, YU Qianpeng, LI Xueyang, ZHANG Zhikai, HU Baigeng. Species and Pathogenicity Identification of Streptomyces Species Causing Potato Common Scab in Ulanqab Area [J]. Chinese Agricultural Science Bulletin, 2024, 40(32): 151-156. |
| [9] | GAO Kaili, MA Xueyan, WANG Haihua, LI Han, LI Yanhua, JIN Wu, WEN Haibo. SNP Analysis of Genetic Diversity and Genetic Differentiation of Whitmania pigra Populations [J]. Chinese Agricultural Science Bulletin, 2024, 40(29): 144-149. |
| [10] | LI Huapeng, PENG Xiaohe, LIANG Xiao, TANG Mingxia, WANG Kexiu, HU Jianjun. Regulation Effect of StGID1 Gene on Potato Plant Height, Diameter and Chlorophyll Content [J]. Chinese Agricultural Science Bulletin, 2024, 40(15): 102-109. |
| [11] | SUN Baojuan, LI Tao, YOU Qian, GONG Chao, LI Zhenxing, LI Zhiliang. Research Progress on MYB Transcription Factors Related to Anthocyanin Synthesis in Solanaceae [J]. Chinese Agricultural Science Bulletin, 2023, 39(36): 102-111. |
| [12] | SUN Bo, LIU Run, WANG Zhanbin, CHEN Huangxin, YAN Su. The Powdery Mildew on Polygonum persicaria L.: Microscopic Observation and Phylogenetic Relationship Analysis [J]. Chinese Agricultural Science Bulletin, 2022, 38(9): 130-136. |
| [13] | DONG Yuqing, WEI Xueping, QIANG Tingyan, ZHANG Bengang, QI Yaodong, LIU Haitao. The Reduced-representation Genome Sequencing Technology: Application in Plant Genetic Analysis [J]. Chinese Agricultural Science Bulletin, 2022, 38(8): 25-32. |
| [14] | CHEN Dao, WANG Xin, JIANG Shan, ZHANG Jie, WU Zujian, DING Xinlun. Strawberry Mottle Virus Isolated in Fujian: Complete Genome Sequence and Molecular Variation [J]. Chinese Agricultural Science Bulletin, 2022, 38(6): 94-101. |
| [15] | Nuerxiati·Nuermaimaiti , LI Huanyu, YANG Chengde. Antagonistic Streptomyces Against Pepper Root Rot Pathogen (Fusarium oxysporum): Screening and Phylogenetic Analysis of 16S rRNA Gene Sequence [J]. Chinese Agricultural Science Bulletin, 2022, 38(33): 116-123. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||